The fundamental role of the retinal pigment epithelium (RPE) is to provide a barrier between the retina and the underlying choroidal blood supply, thus maintaining retinal homeostasis. It is therefore safe to presume that disruption to this protective barrier will cause profound damage to the underlying retina. This article will look at the effects upon the RPE of various pharmaceutical agents commonly used in treatment of disease.
The RPE
Although it is in the periphery, the retina is considered to be part of the central nervous system (CNS) due to the mechanisms of its development.1 In embryonic development (figure 1), the neural plate differentiates into the neural tube and neural crest.

Figure 1: The development of the neural tube (1), the formation of the optic vesicles of each eye (2) and the eventual formation of the retina (3).
The optic vesicle arises from the diencephalon which represents the anterior portion of the neural tube. This vesicle folds in upon itself, forming the optic cup. The retina is the inner layer of this cup, whereas the RPE (figure 2) is the outer layer.
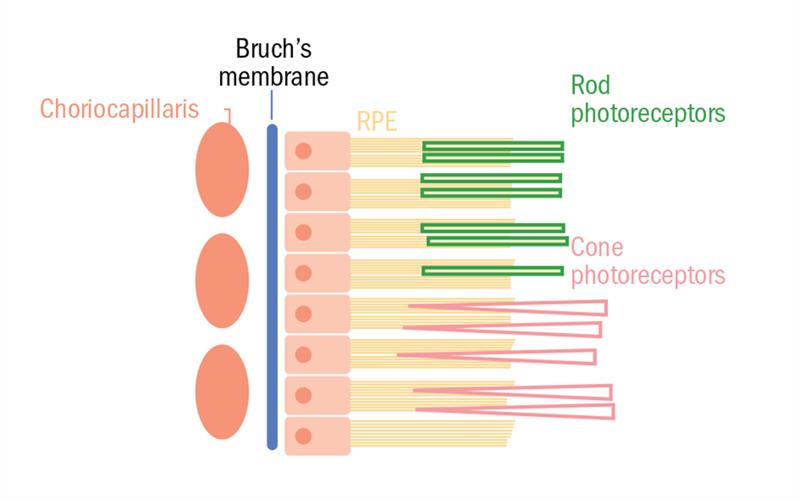
Figure 2: Schematic of the RPE with Bruch’s membrane (and underlying choriocapillaris) and the photoreceptor layer at its basal and apical surfaces respectively
The RPE improves image quality by absorbing stray light2 via a number of pigments (eg melanin, lipofuscin) which absorb light of different wavelengths3. It also has a role in the visual cycle (figure 3). Light absorption in the photoreceptors changes 11-cis retinal into all-trans retinal. Due to the lack of cis-trans isomerase in the photoreceptors, all-transretinal is converted to all-transretinol and transported to the RPE. Here, the cis-trans isomerase re-isomerizes the retinol to 11-cis retinal and this is transported back to the photoreceptor.3

Figure 3: Diagram illustrating the various roles of the RPE in establishing retinal homeostasis. Key: PEDF=pigment epithelium-derived growth factor, VEGF=vascular epithelium growth factor, MV=microvilli, OS=(photoreceptor) outer segment.4
Water, ions and metabolic waste materials are transported across the epithelium from the subretinal space to the choriocapillaris, while glucose and other nutrients are transported from the blood across the epithelium and into the photoreceptors.4 Epithelial transport is not solely responsible for ion homeostasis in the subretinal space. Much like glial cells in potassium spatial buffering, another function of the RPE is responding to rapid changes in the ion composition of the subretinal space, thus maintaining photoreceptor excitability.4 Photoreceptor excitability is also maintained via phagocytosis of photoreceptor outer segments (OS). To avoid the damaging impact of accumulating OS components, the post-mitotic RPE destroys these on a daily basis.5
The RPE secretes growth factors that play a supportive role in photoreceptors and the endothelium of the choriocapillaris.4 As it is derived from the neuroectoderm, the RPE, like the brain, is thought to have a role in immune privilege via the secretion of immunosuppressive factors.4,6
The retinal pigment epithelium (RPE) is a single layer of cuboidal cells which are hexagonal in cross-section, and are a constituent of the outer blood-retinal barrier (oBRB).7 The apical surface of the RPE forms microvilli which envelop the outer segments of photoreceptors, while the basal surface is in contact with Bruch’s membrane which separates the RPE from the choriocapillaris. This morphology accounts for the RPE’s involvement in photoreceptor outer segment renewal, absorption of stray light and removal of free radicals.8 The fundamental role of the RPE, however, is to provide a barrier between the retina and underlying choroidal blood supply, thus maintaining retinal homeostasis.
Harmful xenobiotics
A xenobiotic is defined as ‘a chemical compound foreign to a given biological system’ and includes many therapeutic drugs.9 RPE transporters of naturally occurring substrate may also be receptive to xenobiotics which have similar structures. Therefore, it is possible for certain therapeutic agents to have deleterious effects on the retina, as a result of oBRB damage. Let us now look at four such agents and their negative effects on the retina.
Tamoxifen
Tamoxifen is a commonly used non-steroidal drug prescribed to men and women with the invasive (most common) form of breast cancer after they have had surgical/radiotherapy treatment.10 It is also prescribed to women who have been treated for the less common ductal carcinoma in situ (DCIS), as there is a higher chance of them developing the invasive form later on. It is occasionally given to stimulate ovulation.11
The target of the anti-oestrogen tamoxifen is the oestrogen receptors (ORs) in breast cancer tissue. This is a ligand-activated transcription factor which oestrogen binds to in the cytoplasm. Once bound with oestrogen, the OR is dissociated from heat shock proteins and undergoes phosphorylation and dimerisation before binding to oestrogen response element (ORE) which is found upstream of oestrogen-dependent genes. The transcription of oestrogen regulated genes is altered as a result of OR-ORE binding, and there is up-regulation of positive proliferation regulators via the transcription domains, namely Activating Function 1 and 2 (AF1 and AF2). Tamoxifen-OR binding inhibits activation of AF2 and is an antagonist for genes which it regulates.12
The Adjuvant Tamoxifen: Longer Against Shorter (Atlas) group carried out a longitudinal study which involved 12,894 female subjects who had previously completed five years of tamoxifen therapy for OR-positive early breast cancer. The subjects were randomly allocated into two groups; the first continued therapy for 10 years whilst the second stopped after five years. Entry was between 1996 and 2005 and in 2013 results were reported in a follow-up of 6,846 appropriate (OR-positive) subjects. The results showed that continuing tamoxifen therapy for 10 years further reduced mortality rates from the disease. Future treatment therefore may involve longer term drug use and this would increase the likelihood of tamoxifen induced ocular toxicity. Ocular toxic effects include corneal toxicity, cataract progression, optic neuropathy and macula oedema,13 and tamoxifen retinopathy which is the most significant in terms of affecting vision.14
Kim et al’s study14 proved that tamoxifen toxicity of RPE was dose dependent. Exposing ARPE-19 and fh-RPE (primary foetal human RPE) cells to increasing tamoxifen concentrations led to increased release of lactate dehydrogenase (LDH), indicating lysosomal destabilisation (figure 4). There was also an effect on the cytoskeleton, ie loss of actin myofilaments and ZO-1 proteins of the RPE which disrupted junctions including TJs.

Figure 4: Increasing the dose of tamoxifen over a two-hour period led to a correlating increase lactate dehydrogenase (LDH) release.14
In ARPE-19 cells, initially lysosomal destabilisation occurred, resulting in cathepsin (proteases) release. Cathepsins initiate multiple cell death mechanisms; pyroptosis, apoptosis and necroptosis.14 In apoptosis (programmed cell death), cathepsins within the cytoplasm activate the BH3 interacting-domain agonist (BID) which is a pro-aptotic member of the Bcl-2 group of proteins. This in turn triggers the caspase-9 intrinsic pathway for apoptosis. Necroptosis involves activation of receptor interacting protein 1 (RIP1) and RIP3 kinase. Tamoxifen toxicity is characterised by crystalline deposits within the neural retina. Deposits are also found in the RPE and these contribute to lysosomal destabilisation and therefore activation of NLRP3 inflammasome in macrophages. This activates caspase-1 and hence production of interleukin-1 ß (IL-1 ß) which causes pyroptosis.14
Once visual acuity has been affected in patients with tamoxifen toxicity, the effects are irreversible. Therefore, it is important to monitor patients closely. Ideally practitioners should take baseline measurement of visual acuity, Amsler, central field screening and fundus photography before the patient commences tamoxifen therapy, followed by annual checks.15
Digoxin
Digoxin is a cardiac glycoside commonly prescribed to patients who have cardiac arrhythmia. Though rare, visual disturbances may occur including blurred vision, central scotoma, glare and colour vision defects.16
The membrane bound Na+/K+-ATPase pump is found mainly in cardiac muscle cells. Digoxin works by inhibiting the action of subunits of this pump (figure 5) and in doing so promotes Na+-Ca2+ exchange. Ca2+ influx leads to increased availability of the ion to the contractile elements of the cell thus a greater force of contraction.17
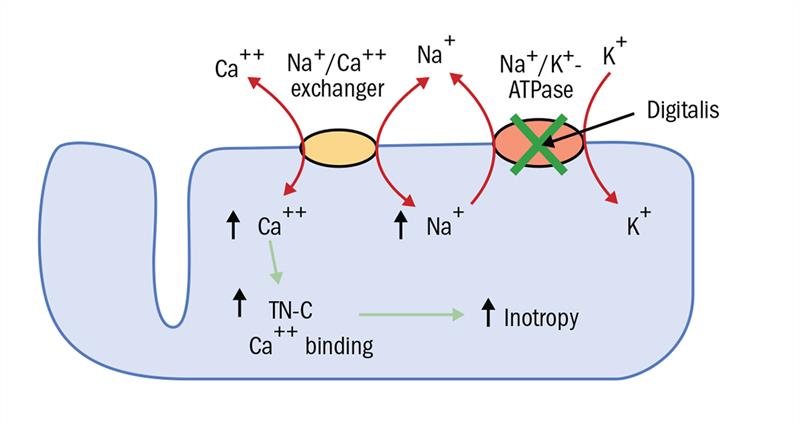
Figure 5: The action of digitalis (digoxin) on a cardiac myocyte in the treatment of cardiac arrhythmia/heart failure. KEY: TN-C= contractile protein where Ca2+ binding occurs. Inotropy= increased muscle action18
Digoxin is a substrate for P-Gp and therefore its concentration in the neural retina is regulated by the RPE. Because of its narrow therapeutic index, care should be taken when prescribing the drug to patients who are also taking P-Gp inhibitors (eg verapamil, quinidine, amiodarone) to avoid toxicity.19 Various case studies have shown that discontinuation of digoxin therapy will resolve the symptoms of toxicity.20,21,22
At doses of greater than ~3nmol/ml, digoxin begins to cause toxic effects. At this concentration, the drug affects the main three subunits of the Na+/K+-ATPase. This causes an overload of intracellular Ca2+ and K+ imbalance; the sarcoplasmic reticulum of the myocardial cells are unable to store any more calcium resulting in an increased Ca2+ inward current and therefore over-contraction of the cardiac muscle, ie cardiac arrhythmias, atrial systolic tachycardia and atrioventricular block.
The ocular side effects of digoxin include colour vision defects, both tritan and red-green deficiency. Lawrenson et al’s study16 constituted of rapid screening tests for tritan and red-green deficiency being carried out on 30 elderly hospital patients who were receiving digoxin treatment. This cohort study observed 30 digoxin therapy patients (10 males and 20 females with a mean age of 81.3 years) and a control group of 30 age-matched hospital inpatients not receiving digoxin (eight males and 22 females with a mean age of 76.8 years). The mean serum concentration of the drug was 1.4nmol/l so no patient had the clinically defined toxic level of digoxin (~3nmol/ml). There were no subjective complaints about colour vision from digoxin patients. Results found that 20 to 30% of patients had a mild to moderate red-green deficiency while ~20% had a tritan deficiency. Therefore, therapeutic doses of digoxin caused impaired colour vision, compared to an age matched control group (figure 6).

Figure 6: Even in the therapeutic dose of the drug, digoxin was capable of causing impairment of colour vision
The colour vision deficiency probably occurs due to digoxin toxicity of a Na+/K+-ATPase found on cone photoreceptors23 although the exact mechanisms are not understood.16 Similar pumps have also been found in Muller cells and RPE.
Chloroquine
Chloroquine and its derivative hydroxychloroquine are anti-malarials that are also commonly used in the treatment of rheumatoid arthritis (RA) as well as other rheumatic diseases, such as systemic lupus erythematosus, sarcoidosis, dermatomyositis, Sjögren’s syndrome, chronic juvenile arthritis and psoriatic arthritis.24
RA is an autoimmune disease (figure 7) in which inflammatory mediators such as tumour necrosis factor-a (TNF- a), IL-1 and IL-6 cause activation of synovial cells and destruction of joints. In 2006 a study by Jang et al showed that chloroquine works by inhibiting release of TNF- a by preventing TNF- a precursor maturation, and decreases levels of IL-1ß and IL-6 mRNA via destabilisation.25

Figure 7: Diagram showing the various inflammatory cytokines released by immune cells in a rheumatoid arthritis patient’s joint
The drugs can cause toxicity in both the cornea and at the macula (resulting in bull’s-eye maculopathy). Corneal toxicity presents as keratopathy which may be a precursor to retinal effects. Chloroquine disrupts the pH of RPE and retinal neural cells and interferes with lysosomal function.26 It affects RPE cell metabolism and therefore causes degradation of photoreceptors, as the RPE’s role in photoreceptor outer segment phagocytosis is compromised. This leads to a breakdown of the oBRB.27 It binds readily to melanin found in the RPE and is eliminated slowly. Pathogenesis of chloroquine-induced retinopathy is hypothesised to be due to the cumulative effect of lipid complexes in retinal neurones and glial cells, resulting in necrotic cell death.28
Bull’s-eye maculopathy presents as a bilateral pigment mottling in the macular region with foveal sparing.15 As a result, there can be irreversible sight loss. Hydroxychloroquine is less likely to cause OARs and is more commonly used in RA treatment. Patients taking the drug should be monitored annually by an optometrist who should carry out central visual fields assessment, colour vision tests, fundus photography and Amsler screening using the red on black grid. The patient should also be able to read small print with the necessary near correction worn in order to start the treatment.15
Vigabatrin
Vigabatrin is an anti-epilepsy drug. Epilepsy is characterized by recurrent seizures which are due to altered electrical signalling within the CNS.28 Synapses at which there is ?-aminobutyric acid (GABA) neurotransmitter activity constitute 30% of all those in the CNS. This is where vigabatrin has its mechanism of action.29 GABA is an inhibitory neurotransmitter and hyperpolarises the membrane of postsynaptic neurones by opening
Cl- channels (via GABAA receptor binding) and K+ channels through a second messenger pathway (via GABAB receptor binding).28 Vigabatrin inhibits the action of GABA transaminase (GABA-T), as seen in figure 8, which is responsible for the breakdown of GABA, thus increasing whole brain levels of the neurotransmitter.28,29

Figure 8: Schematic illustration of an inhibitory synapse and the various components acting at the location. GABA-T normally converts GABA into succinic semi-aldehyde. Vigabatrin inhibits GABA-T found within the pre-synaptic terminal and nearby glial cells.29 Key: GAD= glutamic acid decarboxylase. GAT1= GABA Transporter 1.
Vigabatrin causes bilateral irreversible constriction of VFs in 30 to 50% of patients.15 Most patients with vigabatrin attributed VF constriction (VAVFC) are asymptomatic unless central vison is affected. Field loss is usually concentric and nasally positioned. Electroretinogram (ORG) recordings of VAVFC study subjects are abnormal (figure 9) showing altered ß-waves and a reduction in oscillatory potentials.30

Figure 9: Scotopic ORG response seen in control and vigabatrin (VGB) treated rats. The b-wave is seen to reduce in amplitude in the VGB rat. Oscillatory potentials seen on the b-waves’ ascending limbs are reduced in the VGB rat.31
The exact mechanisms of vigabatrin retinal toxicity are yet to be elucidated, however it is known that vigabatrin inhibition of GABA-T results in GABA accumulation in the retina. This increased concentration is thought to cause excitotoxicity via GABAC receptor activity32 and therefore cell death of retinal neurones that express the receptor, ie cones and bipolar cells.31 It is also thought that light exposure could possibly increase toxicity.32 Vigabatrin was found to cause disorganisation of cell morphology, with photoreceptor nuclei having translocated adjacent to the RPE as one of the manifestations.31 A cohort study by Westall et al33 in 146 children with a form of epilepsy called ‘infantile spasms’ showed that these morphological changes may present in an ORG as early reversible toxicity due to altered cell metabolism, which precedes irreversible defects.
Patients recommended for vigabatrin therapy should have baseline VF screening before commencing treatment. This should be followed up by VF checks every six months. If no defect is seen after five years of therapy, annual monitoring is advised.15
Conclusion
The RPE is a vital component of the BRB. Its many proteins and array of junctions between adjacent cells provide an effective barrier between the retina and the choroidal circulation, maintaining homeostatic conditions in this region.
Due to its importance in terms of consequences to the retina, there has been substantial research into the structure and function of the RPE. The RPE cell lines ARPE-19, D407 and h1RPE have been used widely by researchers who wish to study the morphology of tight junctions and P-Gp within cultured RPE. These cell lines have also proved useful in identifying the effects of various xenobiotics on the RPE in vitro.
The vast majority of studies into the toxic effects of the xenobiotics mentioned in this review have been carried out in RPE cell cultures or animal models. There have also been some studies into the long term effects of the drug on the retina in human subjects, through the use of prospective observational cohort studies and retrospective case studies. For more reliable models of toxin effect in humans there should be further studies of a higher level, ie more observational cohort studies in which an age matched control group is used.
Literature reviewed in this paper has shown that tight junctions and P-Gp can be susceptible to damage from therapeutic drugs which must be taken to treat in some cases life threatening diseases. The extent of this damage may depend on the dose or duration over which the treatment has continued. The mechanisms of certain xenobiotic pathology may involve cell death of the RPE/retinal neurones, but that of others is still to be proved and further research is necessary. Toxicity may be reversible or its progression may be limited if monitored and managed correctly.
References
1. Purves, D; Augustine, GJ; Fitzpatrick, D; Katz, LC; LaMantia, A; McNamara, JO; Williams, SM (eds) (2001) Neuroscience. 2nd Edition. Sunderland (MA). Sinauer Associates
2. Strauss, O. (2005). The Retinal Pigment Epithelium in Visual Function. Physiological Reviews, 85 (3), 845
3. Simó, R; Villarroel, M; Corraliza, L; Hernández, C; Garcia-Ramírez, M. (2010). The Retinal Pigment Epithelium: Something More than a Constituent of the Blood-Retinal Barrier—Implications for the Pathogenesis of Diabetic Retinopathy. Journal of Biomedicine and Biotechnology, 2010, 119-129
4. Strauss, O. (2005). The Retinal Pigment Epithelium in Visual Function. Physiological Reviews, 85 (3), 846
5. Streilein, JW; Ma, N; Wenkel, H; Ng, TF; Zamiri, P. (2002). Immunobiology and privilege of neuronal retina and pigment epithelium transplants. Vision Research, 42(4), 487-495
6. Boulton, M & Dayhaw-Barker, P. (2001). The Role Of The Retinal Pigment Epithelium: Topographical Variation And Ageing Changes. Eye, 15(3), 384-385
7. Miller-Keane & O’Toole, MT. (2005). Miller-Keane Encyclopedia and Dictionary of Medicine, Nursing, and Allied Health. 7th Edition, Saunders
8. Sporn, MB & Lippman, SM. (2003). Agents for Chemoprevention and Their Mechanism of Action, In: Kufe, DW Pollock, RE Weichselbaum, RR Bast, RC Gansler, TS Holland, JF Ill, EF (eds). Holland-Frei Cancer Medicine, 6th Edition. Hamilton
9. Ogbru, O & Marks, JW. (17/02/2015). Tamoxifen. [Online]. Available from: http://www.medicinenet.com/tamoxifen/article.htm [Accessed 29th March 2015]
10. Ring, A & Dowsett, M. (2004). Mechanisms of tamoxifen resistance. Endocrine-Related Cancer, 11(4), 643-658
11. Constable, PA. (2010). Drug Toxicity, MSc Clinical Optometry lectures, City University London
12. Kim, LA; Amarnani, D; Gnanaguru, G; Tseng, WA; Vavvas, DG; D’Amore, PA. (2014). Tamoxifen Toxicity in Cultured Retinal Pigment Epithelial Cells Is Mediated by Concurrent Regulated Cell Death Mechanisms. Retinal Cell Biology, 55(8), 4747-4758
13. Edgar, D. (2014). Principles of toxicology/ Adverse drug reactions, Eye Disease and Therapeutics Lectures, City University London
14. Lawrenson, JG; Kelly, C; Lawrenson, AL; Birch, J. (2002). Acquired colour vision deficiency in patients receiving digoxin maintenance therapy. The British Journal Of Ophthalmology, 86(11), 1259-1261
15. Gheorghiade, M; Adams, KF; Colucci, WS. (2004). Digoxin in the Management of Cardiovascular Disorders. Circulation, 109(24), 2959-2964
16. Klabunde, RE. (12/03/2012). Cardiac Glycosides (Digitalis Compounds). [Online]. Available from: http://www.cvpharmacology.com/cardiostimulatory/di... [Accessed 30th March 2015]
17. Bauman, JL; DiDomenico, RJ; Galanter, WL. (2006). Mechanisms, Manifestations, and Management of Digoxin Toxicity in the Modern Era. American Journal Of Cardiovascular Drugs: Drugs, Devices, And Other Interventions, 6(2), 77-86
18. Hobley, A & Lawrenson, J. (1991). Ocular adverse effects to the therapeutic administration of digoxin. Ophthalmic & Physiological Optics, 11(4), 391-393
19. Butler, VP Jr; Odel, JG; Rath, E; Wolin, MJ; Behrens, MM; Martin, TJ; Kardon, RH; Gouras, P. (1995). Digitalis-induced visual disturbances with therapeutic serum digitalis concentrations. Annals Of Internal Medicine, 123(9), 676-680
20. Wolin, MJ. Digoxin visual toxicity with therapeutic blood levels of digoxin. American Journal Of Ophthalmology, 125(3), 406-407
21. Patel, M. (28th June 2002). Ocular side effects of systemic drugs: Cardiac drugs Part 3. Optometry Today. pp. 34-36
22. Shinjo, SK; Maia Júnior, OO; Tizziani, VAP; Morita, C; Kochen, JAL; Takahashi, WY; Laurindo, IM. (2007). Chloroquine-induced bull’s eye maculopathy in rheumatoid arthritis: related to disease duration? Clinical Rheumatology, 26(8), 1248-1253
23. Jang, CH; Choi, JH; Byun, MS; Jue, DM. (2006). Chloroquine inhibits production of TNF-a, IL-1b and IL-6 from lipopolysaccharide-stimulated human monocytes/macrophages by different modes. Rheumatology, 45(6), 703-710
24. Mahon, GJ; Anderson, HR; Gardiner, TA; McFarlane, S; Archer, DB; Stitt, AW. (2004). Chloroquine causes lysosomal dysfunction in neural retina and RPE: implications for retinopathy. Current Eye Research, 28(4), 277-284
25. Sundelin, SP & Terman, A. (2002). Different effects of chloroquine and hydroxychloroquine on lysosomal function in cultured retinal pigment epithelial cells. APMIS, 110(6), 481-489
26. Ängehagen, M; Ben-Menachem, E; Rönnbäck, L; Hansson, E. (2003). Novel Mechanisms of Action of Three Antiepileptic Drugs, Vigabatrin, Tiagabine, and Topiramate. Neurochemical Research, 28(2), 333-340
27. Ben-Menachem, E. (2011). Mechanism of action of vigabatrin: correcting misperceptions. Acta Neurologica Scandinavica, 124(192), 5-15
28. Hawker, MJ & Astbury, NJ. (2008). The ocular side effects of vigabatrin (Sabril): information and guidance for screening. Eye, 22(9), 1097-1098
29. Duboc, A; Hanoteau, N; Simonutti, M; Rudolf, G; Nehlig, A. (2004). Vigabatrin, the GABA Transaminase Inhibitor, Damages Cone Photoreceptors in Rats. Annals of Neurology, 55(5), 695-705
30. Heim, MK & Gidal, BE. (2012). Vigabatrin-associated retinal damage- potential biochemical mechanisms. Acta Neurologica Scandinavica, 126(4), 219-228
31. Kevany, BM & Palczewski, K. (2010). Phagocytosis of Retinal Rod and Cone Photoreceptors. Physiology, 25(1), 8-15.
32. Bonilha, VL. Retinal pigment epithelium (RPE) cytoskeleton in vivo and in vitro. Experimental Eye Research. [Epub ahead of print: Sep 30, 2013] Accessed at: http://0-www.sciencedirect.com.wam.city.ac.uk/scie...
33. Westall, CA; Wright, T; Cortese, F; Kumarappah, A; Snead III, OC; Buncic, JR. (2014). Vigabatrin retinal toxicity in children with infantile spasms. Neurology, 83(24), 2262-2268.
Dr Paul Constable is a senior lecturer in optometry at Flanders University and Mariyah Mahmood is currently completing her pre-registration qualification