There are many macular conditions that may be encountered by optometrists in practice. They vary greatly in their causes, age of onset, available treatments and symptoms. A number of them will be discussed here
Stargardt disease
Stargardt disease is the most common type of juvenile macular dystrophy. It is a genetic condition, most commonly with an autosomal recessive mode of inheritance (a genetic mutation present in both copies of the gene, in this case the ABCA4 gene) although autosomal dominant cases have been reported. It has an estimated prevalence of one in 10,000. Hundreds of different mutations in this gene can result in this disease or others including cone-rod dystrophy (CRD), Retinitis pigmentosa (RP) and an increased susceptibility to age-related macular degeneration.1,2 It has been shown that one in 20 people are carriers (have a mutation in one copy of their ABCA4 gene).1 The ABCA4 gene codes for a membrane protein, an ABC transporter situated in the disc rims of rod and cone photoreceptor outer segments.
Its role involves utilising adenosine triphosphate (ATP) in the removal of the toxic all-trans form of oxidised vitamin A (retinol) from photoreceptors after it has stimulated the visual transduction pathway. Otherwise the all trans form reacts to form a compound called N-retinylidene-phosphatidylethanolamine (N-retinylidene-PE) which can also be transported by ABCA4.3 In Stargardt disease transport is not as efficient, enabling an extra molecule of all trans retinal to bind to N-retinylidene-PE creating a compound called A2PE.4 Old receptor outer segments are phagocytosed and A2PE broken down into A2E by lysosomal enzymes in the retinal pigment epithelium (RPE). A2E is not easily metabolised and builds up in RPE cells, resulting in lipofuscin accumulation. This in turn can result in RPE cell death and atrophy of overlying photoreceptors.
The onset of Stargardt disease usually begins in the first two decades of life, although there is a wide range of severity. If it manifests later in life, progression tends to be slower (such as a phenotype known as fundus flavimaculatus). Individuals may be aware of a bilateral deterioration and distortion in their central vision, especially in dim lighting and complain of difficulty with reading.5 The loss of visual acuity is bilateral and progressive and usually results in a final Snellen acuity of between 6/60 and 6/120.6 Patients may have a mild red-green colour defect at early stages that gradually progresses.7
At the preliminary stage of Stargardt disease the macula often appears normal and the macular reflex may even be present. In early stages there is usually only a subtle granular mottling effect at the fovea.8 In some individuals a ring of approximately one disc diameter develops at the level of the RPE encompassing the fovea. It is comprised of flecks that may appear similar to drusen but with a variety of shapes including a fish tail appearance (pisciform). However, the extent and location of flecks can also vary greatly between patients with some not developing any at all.9 They occur peripherally in fundus flavimaculatus in contrast to Stargardt.
Various electrophysiological tests can be used to aid diagnosis of Stargardt disease and other macular dystrophies. The electoretinogram measure changes in the electric potential of the eye following presentation of a given stimuli and shows typical patterns in dark (scotopic) and light (photopic) conditions. This enables the cone, rod receptor and ganglion cell function to be assessed.10 The electro-oculogram (EOG) is usually an adjunct to the ERG. It measures the standing potential between the electrically positive cornea and the electrically negative back of the eye (Bruch’s membrane) during adaptation to light and dark environments. An abnormal EOG arises as a result of problems in the RPE.10 As Stargardt disease develops, the EOG and ERG measurements that tend to be normal in the initial stages begin to indicate mildly reduced rod, or rod and cone responses or impaired dark adaptation respectively.9 The EOG is usually normal until the advanced stages.11 The stage at which this develops is variable, reflecting the disease spectrum.
Fleck reabsorption and extensive choriocapillaris atrophy occur together with RPE atrophy observed in advanced cases. Significant RPE atrophy results in geographical atrophy at the macula, with a beaten bronze or bull’s eye appearance that may develop associated areas of hyper-pigmentation (Figure 1). In advanced cases, visual field defects and significant loss of sensitivity of rods and cones with ERG testing are typical.
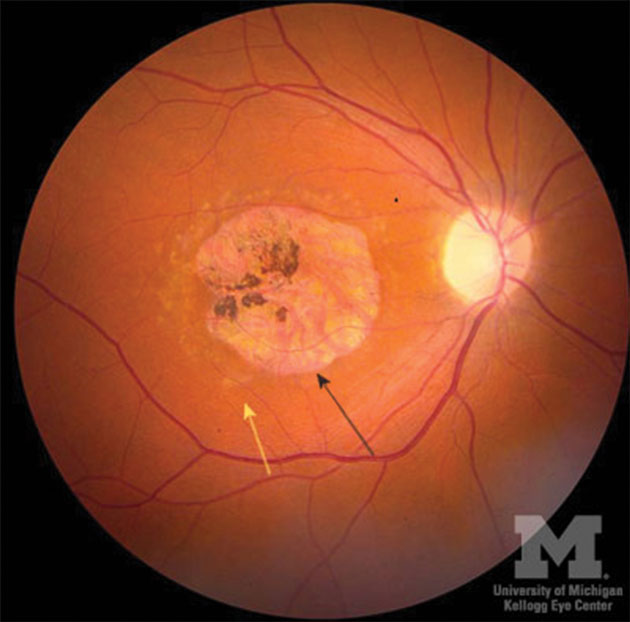
[CaptionComponent="2753"]
Fluorescein angiography (FA) may also be performed to confirm the diagnosis of Stargardt disease. FA is restricted to hospital settings due to the need for resuscitation facilities in the rare event of an anaphylactic reaction. A yellow dye is injected into a vein in the arm and images taken of the retinal vessels as the dye travels through. The resultant patterns of fluorescence can aid diagnosis. Cases of Stargardt disease may be characterised by a dark (hypofluorescent) choroid effect due to blockage or reduction of the background fluorescence by increased lipofuscin in the RPE (Figure 2).13 In areas where there is RPE cell loss due to atrophy, FA produces corresponding hyperfluorescence (window defects).13 This phenomenon does not directly correspond to the flecks seen on ophthalmoscopy.
[CaptionComponent="2754"]
Fundus autofluorescence imaging can also help confirm the diagnosis of Stargardt disease. It enables the visualisation of bisretinoid components (including A2E) of lipofuscin in the RPE. The flecks often visible in Stargardt disease appear hyperfluorescent, whereas RPE atrophy is hypofluorescent.13 Genetic testing may be available but not all patients have a known mutation in the ABCA4 gene and it takes time to produce the result.
During fundoscopy the accumulated lipofuscin results in the fundus appearing a reddish or brown colour. There is a lack of choroidal detail as this is blocked by the overlying lipofuscin.9 Sparing of the peripapillary RPE is also usually observed in Stargardt disease.9
Fundus photography can help establish baseline and monitor any disease progression in practice due to lack of subjective variation between practitioners. Ocular coherence tomography (OCT) provides a cross-sectional image of the retina based on variation in reflective properties of the different layers. If this is available the flecks may be seen as hyper-reflective elements at the level of the RPE on a macular OCT scan.
There may be disruption of the inner segment ellipsoid (ISe) band also known as the PIL (photoreceptor integrity line), the border between the inner and outer photoreceptor segments. Thinning of the photoreceptor layer may also be present. In advanced cases of the disease, thinning of the retinal layers, RPE and enhanced reflectivity with thinning of the choroid can be observed.14 The advantage of OCT, particularly with Stargardt, is that the patient’s retina isn’t being exposed to significant and potentially harmful visible light.
At present there is no mainstream treatment available for Stargardt disease. Use of sunglasses to protect against UV and a cap or hat with a brim to shield the eyes is advisable. Experimental treatments include stem-cell therapy to replace damaged RPE cells. Drug-based therapies in clinical trials include modified vitamin A, which makes it harder for the vitamin A dimer and consequently A2PE to form. This theory is under investigation using mice models with Stargardt mutations. Another approach currently undergoing clinical trials involves the delivery via a vector of a normal ABCA4 gene to photoreceptor cells.15
Cone dystrophies
Cone dystrophies are a group of rare conditions with different modes of inheritance (mainly autosomal dominant, but also autosomal recessive and X-linked).15 They are either stationary (occurring congenitally with consistent symptoms throughout life) or progressive (symptoms worsening with time). This article will concentrate on the progressive group.
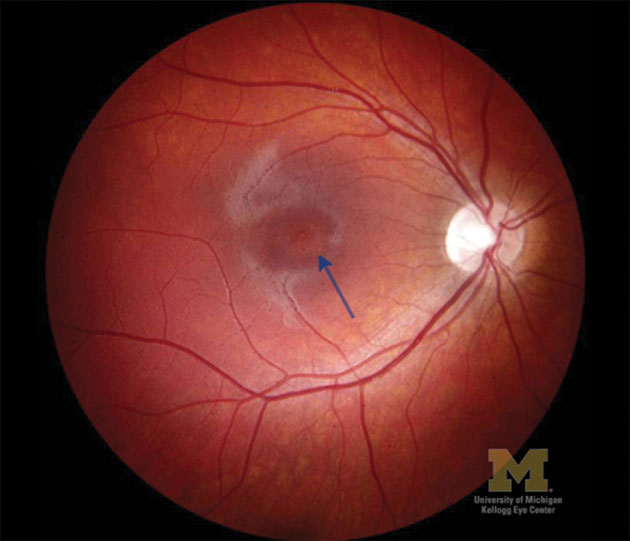
[CaptionComponent="2755"]
Progressive cone dystrophies describe a spectrum of disorders usually presenting in older childhood or early adulthood before the age of 30. They are characterised by symptoms of photophobia and glare and a progressive reduction in visual acuity, although there is a wide spectrum with Snellen acuities ranging from 6/6 to hand movements.16 Individuals also have various colour vision defects (dyschromatopsia) early in the disease process. The fundus often initially appears normal or there may be subtle macular changes or as the condition progresses Bull’s eye maculopathy may develop (as in Stargardt disease) culminating in macular atrophy (Figure 2). Visual field defects (central scotoma) also develop later. Progressive cone dystrophies may be seen in isolation or as an aspect of various syndromes.
Individuals with cone dystrophy have normal rod function, whereas cone-rod dystrophies are also associated with additional symptoms including night blindness (nyctalopia) and peripheral visual field loss that develop later when the rods are also affected. Visual acuity also continues to reduce to the point of legal blindness and nystagmus may develop.
Cone dystrophies are characterised by a severely abnormal or non-recordable photopic ERG. A rod isolated ERG will produce normal or almost normal results and the patient’s peripheral visual fields will be full in contrast to an individual with advanced cone-rod dystrophy (in whom cone fuction is more severely affected than rod).17
In cone-rod dystrophies, fundus signs progress from bilateral macular atrophy and optic disc pallor and narrowing of retinal vessels may occur later.18 Unfortunately, there is no treatment. Confirmation of diagnosis of these conditions and genetic counselling can be helpful. The patient should be referred to their GP routinely for further ophthalmological assessment.19
The patients with the conditions discussed are all likely to develop a central scotoma. All these patients are young and such changes have the potential to have a significant impact on their education. A low vision assessment including eccentric viewing training may help maximise remaining vision. However, as the distance of the new retinal area being using (preferred retinal locus or PRL) from the macular increases the maximum visual acuity that can be achieved decreases. To compensate for this, magnification is required. The low vision assessment may also involve training in the use of and trialling various types of optical magnifiers. Software is also available to increase computer screen text size or show single words on the screen.20,21
Referral can also help confirm diagnosis pinpointing the exact condition and enabling the individual and possibly their family more information regarding visual prognosis. Blind or partially sighted registration can be arranged by the ophthalmologist, if the patient is eligible and in agreement with the issuing of a certificate of visual impairment (CVI). Processing of this form enables individuals to register with their local social services that can entitle them to various allowances and facilitates the process of claiming applicable benefits.22 Following diagnosis, people with these conditions often understandably require support.
The patient’s GP may be able to arrange counselling.19 They can also be given details of various charities to follow up if they wish. The Royal National Institute of Blind People (RNIB) provides a telephone helpline and an emotional support service.21 The Macular Society arranges local support group meetings as another means of assistance.21 It is also advisable to recommend that family members have eye examinations due to the genetic mode of inheritance of these conditions. Although there is currently no treatment for these diseases, it is important that these individuals continue to have regular eye examinations so that if they develop any other eye conditions such as diabetes, glaucoma or cataract they can be diagnosed and treated promptly to prevent their remaining vision being compromised.
Cystoid macular oedema (CMO)
CMO is characterised by fluid accumulation in cystic spaces in the outer plexiform and inner nuclear retinal layers. This can often be visualised using a macular OCT scan if available (Figure 4).
[CaptionComponent="2756"]
It is most commonly observed following ocular surgery. Anterior ocular inflammation resulting from surgery triggers the release of endogenous inflammatory mediators. Prostaglandins, cytokines, and other factors increase the permeability of perifoveal capillaries, culminating in the breakdown in the blood-retinal barrier. CMO changes include an increase in retinal thickness and loss of foveal depression. During indirect slit-lamp ophthalmoscopy it may be possible to see a subtle elevation at the macula. The patient may have a uniocular hyperopic shift compared to previous refractions.
CMO is also associated with various other systemic and eye conditions. These include circulatory disorders including diabetes and vascular occlusion, ocular inflammation, epiretinal membranes, genetic disorders (retinitis pigmentosa, autosomal dominant CMO) and drug toxicity (nicotinic acid). Patients may be asymptomatic or may notice a reduction in visual acuity.
Topical non-steroidal anti-inflam-matory eye drops (NSAIDS) such as bromfenac are prostaglandin inhibitors and often the first line treatment. There are other topical NSAIDS as well such as nepafenac, flurbiprofen and ketorolac that may be used as an alternative. If this does not resolve the condition, corticosteroids may be administered via a sub-tenon or subconjunctival injection as NSAIDs and corticosteroid inhibit different aspects of the inflammatory cascade. Other methods of administration for persistent CMO include an intravitreal injection of corticosteroid such as triamcinolone acetonide or anti-VEGF such as bevacizumab, aflibercept or ranibizumab. VEGF contributes to blood retinal barrier breakdown by increasing capillary permeability.
Tamoxifen maculopathy
Tamoxifen is a non-steroidal oestrogen antagonist drug used to treat oestrogen positive breast cancer. Oestrogen receptors are present in not only the anterior and posterior segments of the eye but also in lacrimal and meibomian glands, so theoretically the potential for ocular side-effects is great. Ocular side-effects may include retinal refractile crystalline deposits that may be observed on fundoscopy and with OCT (usually situated in the nerve fibre and inner plexiform retinal layers close to the fovea). Studies have shown that this occurs between approximately 1 and 6 per cent of breast cancer patients.23 Crystalline maculopathy cases should be referred routinely to their G.P.19 Oedema may also be present and can also be assessed with OCT or FA.
A macular OCT scan may reveal cystoid spaces at the fovea and loss of photoreceptors. FA may produce a window defect as punctate grey lesions on the RPE may block fluorescence and cystoid macular oedema if it is present.24 Oedema may resolve on cessation of the drug, whereas refractile crystals tend to persist.25 Individuals may be asymptomatic or complain of a mild to moderate decline in their visual acuity. Presence of oedema would necessitate a timely referral for ophthalmological assessment via their GP, detailing in the letter the clinical findings and that the patient is currently taking tamoxifen. Copies of fundus photographs and OCT scans should ideally be included. If oedema is not treated, retinal atrophy and a subsequent reduction in visual acuity can result.
A number of trials have also found an increased risk of cataract development either following or during tamoxifen use.26 Corneal vortex keratopathy (corneal whorls most commonly present in the inferior corneal epithelium) has also been associated with tamoxifen.27 Fortunately, lower doses of tamoxifen are now prescribed for breast cancer treatment, so side-effects are less common.
This article has discussed a number of macular conditions, some more common than others but as primary care practitioners we may encounter any if not all of them in our careers. We have a vital role in establishing the initial potential diagnosis and determining the appropriate management, whether it be monitoring in practice or referring for further confirmatory assessment and treatment.
Model answers
(The correct answer is in bold text)
1 What type of colour vision anomaly is associated with Stargardt diseases in the initial stages
A Tritanopia
B Achromatopsia
C Red-green defect
D Tritanomaly
2 Which new cases required fast track referral
A Stargardt disease
B Cone dystrophy
C Crystalline maculopathy
D Central serous retinopathy
3 Cystoid macular oedema may be characterized by:
A Subretinal fluid accumulation
B Beaten bronze effect at the macular
C Hyperopic shift
D Disciform lesions
4 Reported ocular side effects of tamoxifen include
A Crystalline maculopathy
B Cystoid macular oedema
C Optic neuritis
D Retinal vasculitis
5 Hypofluorescence in fundus autofluoresence can indicate the presence of
A RPE atrophy
B Lipofuscin accumulation
C Presence of bisretinoids
D None of the above
6 The ABCA4 receptor is situated in:
A Bipolar cells
B Amacrine cells
C Cone and rod cells
D Ganglion cells
Acknowledgement
Thanks to Associate Professor David Zacks at the University of Michagan for the Stargardt and cone dystrophy images.
References
1 Zernant J, Schubert C, Im KM, Burke T, Brown CM, Fishman GA, Tsang SH, Gouras P, Dean M, Allikmets R. Analysis of the ABCA4 Gene by Next-Generation Sequencing. IOVS, 2011; 52(11):8479-8487.
2 Allikmets, R, Shroyer, NF, Singh, N, Seddon, JM, Lewis, RA, Bernstein, PS, Peiffer, A, Zabriskie, NA, Li, Y, Hutchinson, A, et al Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science, 1997; 277:1805–1807.
3 Travis GH. Photo-transduction: The Visual Cycle p648. In: JC Besharse D Bok, editors.The Retina and its Disorders. Academic Press. 2011.
4 Quazi F, Lenevich S, Molday RS. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nature Communications 3, Article number: 925
5 http://lowvisionmd.org/stargardts-disease/
6 Sherman J. A novel biomarker for Stargardts Disease 2013.
7 Onofrey BE, Skorin L, Holdeman NR. Ocular Therapeutics Handbook A Clinical Manual. 2nd Ed. Lippincott Williams and Wilkins 2005.
8 Vollmer LA, Shechtman DL, Woods AD, Pizzimenti JJ. Use of multifocal ERG and OCT for diagnosing Stargardt disease. Clinical and Experimental Optometry, 2011:309-313.
9 Sohn HN, Mullins RF, Stone EM Chapter 42. Macular Dystrophies. In: Retina SJ Ryan, In: Retina. 5th Ed. Elsevier Saunders 2013; p864-869.
10 Wolpert K, Tsang K. Electroretinography 2011.
11 Margolis S. Electrodiagnosis and Hereditary Retinal Disease 2002. The New York Eye and Ear Infirmary.
12 Zacks D, personal communications, Kellogg Eye Center. University of Michigan.
13 Dithmar S, Holz HG. Chapter 5. Macular Disorders. In: Fluorescence Angiography in Ophthalmology. Springer 2008; p104
14 Puech B, De Lary J J. Chapter 17. Stargardts Disease. In: B Puech, J J De Lary, GE Holder, editors. Inherited Chorioretinal Dystrophies. A text book and Altas. Springer 2014 p185-194.
15 www.oxfordbiomedica.co.uk/stargen/
16 Telander DG, Small KW. Chapter 6. Retina and Vitreous. 6.14. Macular Dystrophies. In: M Yanoff, JS Duker, editors. Ophthalmology. 4th Edition. Saunders Elsevier Saunders 2014: p500.
18 Tsui I, Song B, Lin C, TSang SH, A Practical Approach to Retinal Dystrophies 2007.
19 Benson M. The spotty retina. Optometry, Today 2012:30-34.
20 Trauzettel-Klosinski S, Current Methods of Visual Rehabilitation. Dtsch Arztebl Int. 2011; 108(51-52):871–878.
21 RNIB. Stargardt disease. RNIB 2012.
22 Rotheroe A, Bagwell S, Joy I. IN SIGHT: A review of the visual impairment sector 2013. The Clothworkers’ foundation.
23 Eisner A, Luoh S-W. Breast Cancer Medications and Vision: Effects of Treatments for Early-stage Disease. Curr Eye Res, 2011; 36(10): 867–885.
24 Noffke AS, Jampol LM. Chapter 6. Areas of Fundus Whitening: White or Yellow spots. In: Clinical Pathways in Vitreoretinal Disease. Steidl SM, Hartnett ME. Thieme 2003; p71.
25 Mittra RA, Mieller WF. Chapter 89. Drug Toxicity of the Posterior Segment. In: RP Schachat, SR Sadda editors Retina. 5th Ed Vol 2. Elsevier Saunders 2013; p1532.
26 Visvanathan K, Chlebowski RT, Hurley P, et al. American Society of Clinical Oncology Clinical Practice Guideline Update on the Use of Pharmacologic Interventions Including Tamoxifen, Raloxifene, and Aromatase Inhibition for Breast Cancer Risk Reduction. JCO, 2009; 27(19): 3235-3258.
27 Chaglasian E, Than TP. When Collateral Damage’ Strikes the Cornea. 2013.
Louise Stainer is a hospital optometrist at Optegra Eye Hospital, Birmingham where Salman Mirza is a consultant ophthalmologist