As a practitioner, whether you are an IP optometrist, optometrist, contact lens optician or dispensing optician, it is fair to say that you will likely spend a significant amount of time during your working week discussing dry eye or dealing with dry eye-related issues; whether you realise it or not. Even our support staff - receptionists, clinical assistants or ‘the otherwise named’ will no doubt occasionally be asked about dry eye by the Great British public. Our pharmacist, GP and ophthalmology colleagues are in a similar boat with swathes of patients seeking their input for a variety of eye related ailments, which can often be attributed, however inaccurately, as dry eye.
Whether you work in a practice with a dedicated dry eye clinic, hospital eye clinic, specialist contact lens practice, high street optical chain, independent optical practice or somewhere in between, dry eye patients pose several challenges to your practice from contact lens drop out, spectacle non-tolerance to simply filling our appointment diaries with at times challenging patients. Such patients, in my opinion, offer us an opportunity to gain a patient for life ¬ a rare commodity these days with so much
competition on the high street. If you take the time to actively listen to the patient’s woes, dust off your anatomy and physiology, flex your diagnostic muscles with a detailed and systematic examination, provide some hand-holding and somehow manage to solve or at least ease their symptoms – or in some cases suffering – such patients become your biggest fan and more importantly a strong and active advocate for your practice. You may even enjoy yourself, finding the process professionally rewarding.
In parts one to four of this series we dealt with neuropathic dry eye (NDE) and evaporative dry eye (EDE), offering an overview of the aetiology of each and providing case histories highlighting some of the treatment methodologies available to us as practitioners. Parts five and six will deal with aqueous deficient dry eye (ADDE), the less common but often more challenging of the ‘big two’.
Function of the tear film
The tear film is the primary interface of the eye, situated between the ocular surface and the outside environment. As has been previously discussed, it plays a critical role in the health and optical performance of the eye. Maintenance of the quality and quantity of the tear film requires a dynamic system, consisting of tear production, tear drainage through the nasolacrimal duct, fluid absorption or exchange through the conjunctival and corneal epithelium, and evaporation to the air.1 An adequate volume of tears is an important prerequisite for a healthy and clear ocular surface. A reduction in the volume of tears gives rise to a greater propensity for developing signs and symptoms associated with ocular dryness and thus causing a significant disturbance in the optical surface of the cornea.2
Mechanisms of DED
While great emphasis is placed on correctly identifying and sub-dividing the diagnosis of DED into its various forms and providing some insight into the severity to help guide our treatment advice, it is also important to remember the delicate interplay between different forms of dry eye. A clear diagnosis of ADDE or EDE is not always possible. Simultaneous ADDE and EDE cases are not impossible nor uncommon. EDE patients are likely to dominate your clinic, however, cases of ADDE in my experience are often more challenging to treat and involve some of the most visually at risk patients you will encounter.
As way of a massively simplified summary, the following aims to explain the complex interplay of factors that comprise the drivers of DED depicted in figure 1.
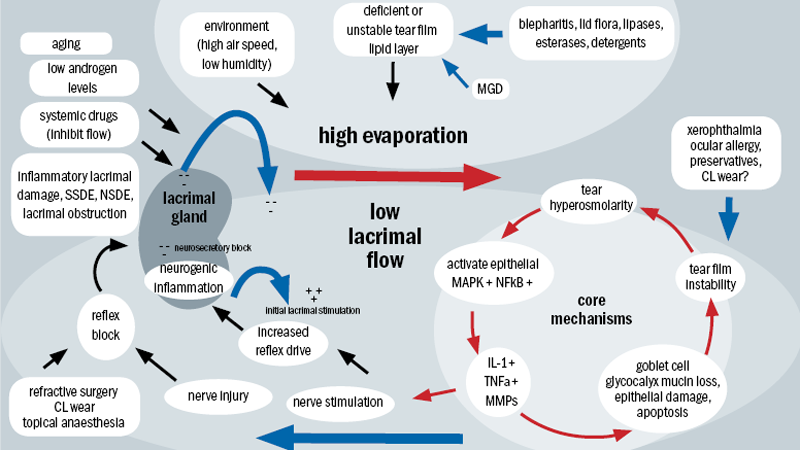
Figure 1: Mechanism of dry eye disease
The viscous cycle – a brief overview
The core mechanisms of dry eye are driven largely by tear film hyperosmolarity and tear film instability.3 The cycle of events in figure 1 outlines and highlights the complexity, the interactions and the plethora of factors that can drive DED.
Tear film hyperosmolarity causes damage to the corneal surface epithelium by initiating a cascade of inflammatory events at the ocular surface, triggering the subsequent release of inflammatory mediators into the tears.4 Corneal epithelial damage involves cell death, as a result of apoptosis, loss of goblet cells and disturbance of mucin expression, leading to tear film instability.5-7 This induced tear film instability in turn exacerbates ocular surface hyperosmolarity, culminating in and completing the viscous cycle.8
Why the symptoms?
DED induced epithelial injury stimulates corneal nerve endings leading to the patient’s symptoms of discomfort, increased
blinking and compensatory reflex lacrimal tear secretion.9 The symptoms are further exacerbated by the increased frictional resistance between the lids and globe as a result of loss of normal tear mucins.10-11
Tear hyperosmolarity
Tear hyperosmolarity is the central mechanism causing ocular surface inflammation, damage, and symptoms, and the initiation of compensatory events in dry eye.12-13 Reduced aqueous tear flow induced by lacrimal failure and/or increased tear film evaporation offer the most likely cause of tear hyperosmolarity.14
Environmental conditions of low humidity and high air flow can cause increased evaporative loss, which can also be caused by meibomian gland dysfunction (MGD).15 The result is an increasingly unstable tear film lipid layer. The impaired delivery of lacrimal fluid into the conjunctival sac causes reduced aqueous tear flow. This can also be induced by certain systemic drugs including antihistamines and anti-muscarinics, but may also be a feature of normal ageing.16 However, inflammatory lacrimal damage, which is seen in autoimmune disorders such as Sjögren’s syndrome dry eye (SSDE) and also in non-Sjögren’s syndrome dry eye (NSSDE), is the most common cause of tear film hyperosmolarity, causing both tissue destruction and neurosensory block.17-20
Reflex block
Various aetiologies may cause dry eye as a result of reflex sensory block, resulting in a loss of sensory reflex drive to the lacrimal gland from the ocular surface.21-24 These include refractive surgery (LASIK dry eye),25-28 chronic use of topical anaesthetics29-30 and contact lens wear.31-37 Cicatricial conjunctival scarring can obstruct tear delivery which, like reflex sensory block, can lead to lower tear delivery.38-40 The chronic surface damage induced by both can lead to reduced corneal sensitivity and thus a reduction reflex tear secretion, this culminates in another vicious cycle.
The overlap – ADDE and EDE
Clinical and anecdotal experience tells us that clinical separation in the diagnosis of ADDE and EDE may in many cases be difficult based on substantive tests. While there are many studies that conclude that, as one would expect, tear evaporation rate is increased in meibomian gland dysfunction (MGD),41-42 in some groups of MGD evaporation rate may be normal.43 The same contradictions can be found within ADDE groups with studies reporting increased evaporation rate44-45 and others reporting decreased evaporation rates.46
Contradictions can also be found in reduced tear flow, a hallmark of ADDE. Numerous studies confirm reduced tear flow in ADDE patients,47 but similar findings have also been reported with MGD patient groups.48 Such contradictions may simply highlight our limited understanding of these conditions, but should also highlight the importance of a systematic unbiased approach to the evaluation of the dry eye patient. There is evidence of reduced or retarded tear film lipid layer spreading in severe ADDE, which has been attributed to the effect of the thinned aqueous phase of the tear film. Is has also been suggested that loss of corneal sensitivity in EDE could reduce the reflex drive to tear secretion and therefore result in a combined form of dry eye. Such interaction occurring over prolonged periods of time may explain the overlap in findings in EDE and ADDE, as explained by figure 1 and the summary above.
Aqueous deficient dry eye (ADDE) – the detail
Aqueous tear-deficient dry eye implies that dry eye is due to the failure of lacrimal tear secretion because of lacrimal acinar destruction or dysfunction, culminating in progressively reduced lacrimal secretion and overall tear volume. As explained earlier this in turn likely causes tear hyperosmolarity. The reasoning behind the change in tear osmolarity is simple – the aqueous evaporates from the ocular surface at a normal rate, however it is evaporating from a reduced aqueous tear pool and therefore leads to hyperosmolarity. ADDE has two major subclasses – SSDE and NSSDE as shown in figure 2.
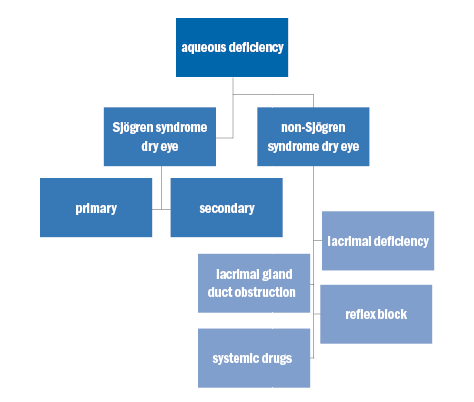
Figure 2: Aqueous deficiency
Sjögren syndrome dry eye (SSDE)
Sjögren syndrome (SS) is an exocrinopathy in which the lacrimal glands, salivary glands and other organs are targeted by an autoimmune process. The resultant chronic inflammation leads to xerostomia (dry mouth) and keratoconjunctivitis sicca in about 95% of patients.49 The lacrimal and salivary glands are infiltrated by activated T-cells which cause acinar and ductular cell death resulting in hyposecretion of the tears and/or saliva.17,50 This hyposecretion is further fuelled by neurosensory block due to the effects of inflammatory cytokines or as a result of circulating antibodies directed against muscarinic receptors within the glands.51 The frequency of MGD is higher in patients with SS than in the normal population.43 Dry eye syndrome with SS is often more severe than non-SS dry syndrome.55-56 Lack of both basic and reflex tearing resulting from lacrimal gland destruction leads to severe ocular surface staining.57
SS can be sub-divided into two forms; primary SS and secondary SS.52
Primary SS
Primary SS consists of ADDE in combination with xerostomia, the presence of autoantibodies, evidence of reduced salivary secretion and a positive focus score on minor salivary gland biopsy.53
Secondary SS
Secondary SS consists of the features of primary SS in conjunction with overt autoimmune connective disease such as rheumatoid arthritis (the most common), systemic lupus erythematosus, Wegener’s granulomatosis, primary biliary sclerosis, systemic sclerosis, polyarteritis nodosa, scleroderma or mixed connect tissue disease.54
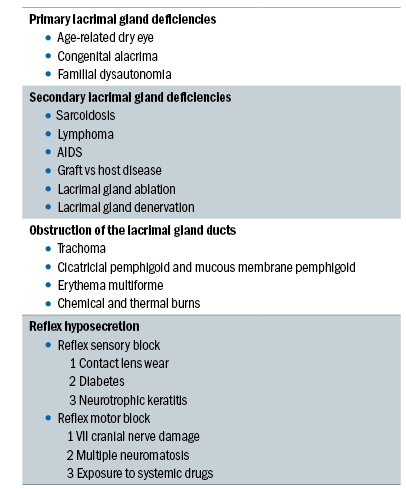 Table 1: Conditions associated with non-Sjögren syndrome dry eye
Table 1: Conditions associated with non-Sjögren syndrome dry eye
Non-sjögren syndrome dry eye (NSSDE)
NSSDE is a form of ADDE caused by lacrimal dysfunction with the important distinction that systemic autoimmune characteristics of SSDE have been excluded. NSSDE can be further sub-divided into four sub-categories (as shown in table 1):
- Primary lacrimal gland deficiencies
- Secondary lacrimal gland deficiencies
- Obstruction of the lacrimal gland ducts
- Reflex hyposecretion
Primary lacrimal gland deficiencies
This most common of the primary lacrimal gland deficiencies is age-related dry eye (ARDE). Studies have shown significant age-related correlations in tear evaporation, volume, flow and osmolarity,58 with further studies showing an age-related relationship in tear turnover,59 tear evaporation60 and lipid layer.61 An increase in ductal pathology that could promote obstructive lacrimal gland dysfunction has been shown with increasing age in the normal human population.62 Periductal fibrosis, interacinar fibrosis, paraductal blood vessel loss and acinar cell atrophy are all thought to be potential causes, with a study reporting lymphocytic glandular infiltrates in 70% of lacrimal glands studied, this is considered to be the basis of the fibrosis.63 Subclinical conjunctivitis is also suggested as being responsible for stenosis of the excretory ducts. Other less common causes of primary lacrimal gland deficiencies include congenital alacrima64-67 and familial dysautonomia.68,69
Secondary lacrimal gland deficiencies
A number of potential causes of secondary lacrimal gland deficiencies have been reported including lacrimal gland infiltration, sarcoidosis, lymphoma, AIDS, graft vs host disease (GVHD), lacrimal gland ablation and lacrimal gland denervation.
In lacrimal gland infiltration, lacrimal secretion may be reduced or fail because of inflammatory infiltration of the gland. Similarly, in sarcoidosis infiltration of the lacrimal gland by sarcoid granulomata70 and in lymphoma infiltration of the lacrimal gland by lymphomatous cells may cause dry eye.71 In autoimmune deficiency syndrome (AIDS) patients, there is a predominance of CD8 suppressor cells, rather than CD4, helper cells leading to DED.72 Dry eye is a common complication in GVHD, occurring around six months after hematopoietic stem cell transplantation as a result of lacrimal gland fibrosis.73,74 Dry eye may be caused by ablation of lacrimal gland at any age or by severance of the ducts, which enter into the superolateral fornix, during lid surgery. Dry eye is not an inevitable outcome since the accessory glands and conjunctiva secretions may compensate in some cases.75 Parasympathetic denervation of the human lacrimal gland may also cause dry eye.76
Obstruction of the lacrimal gland ducts
Obstruction of the ducts of the main palpebral and accessory lacrimal glands leads to ADDE and can be caused by various forms of cicatrising conjunctivitis including trachoma, cicatricial pemphigoid, mucous membrane pemphigoid, erythema multiforme, and chemical and/or thermal burns. In these conditions, conjunctival scarring can also lead to cicatricial obstructive MGD. This effect is seen in trachoma where a combination of tarsal and conjunctival scarring leads to cicatrizing meibomian gland obstruction and trichiasis which causes dry eye as a result of lacrimal obstruction, lid malposition and a deficient tear film lipid later, ultimately leading to corneal opacity and blindness.77 Dry eye caused by lacrimal obstruction, cicatricial MGD, and/or poor lid apposition is also seen in cicatricial and mucous membrane pemphigoid, mucocutaneous disorders characterised by blistering of the skin and mucous membranes, leading to severe and progressive conjunctival scarring.78-80 Conjunctival scarring caused by diffuse chemical and thermal burns, and as a result of erythema multiforme (an acute, self-limiting mucocutaneous disorder usually precipitated by drugs, infection or malignancy) can also all lead to dry eye in this manner.81,82
Reflex hyposecretion
Reflex hyposecretion can be sub-divided into:
- Reflex sensory block
- Reflex motor block.
A summary of the causes of ocular sensory loss are provided in table 2.
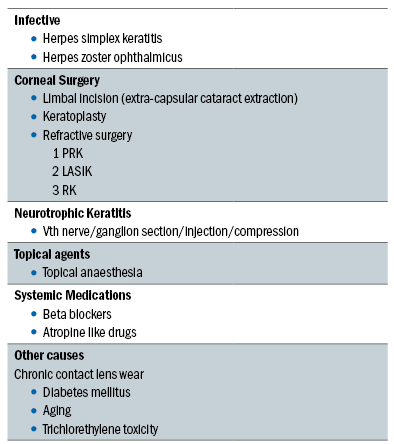
Table 2: Causes of ocular sensory loss
Reflex sensory block
In reflex sensory block, several factors have been shown to be of influence including contact lens wear (hard and extended wear contact lens wear),83 LASIK refractive surgery,84 diabetes mellitus85-88 and neurotrophic keratitis.89 Trigeminal sensory input arising mainly from the nasolacrimal passages and the ocular surface of the eye drives lacrimal tear secretion in the waking state. When the eye is open increased reflex sensory drive occurs from the exposed ocular surface. A reduction in sensory drive from the ocular surface is thought to cause dry eye by decreasing reflex-induced lacrimal secretion and by reducing the blink rate, therefore increasing evaporative loss.90 The conditions listed above can lead to trigeminal denervation and bilateral sensory loss, thus reducing both tear secretion and blink, and therefore leading to DED.91
Reflex motor block
Damage to the VII cranial nerve leads to dry eye due to loss of lacrimal secretomotor function, resultant lacrimal hyposecretion and lagophthalmos. Multiple neuromatosis and systemic drug use have also been noted by several studies to induce dry eye as a result of decreased lacrimal secretion.92 Drugs including antihistamines, beta blockers, antispasmodics, diuretics, tricyclic antidepressants, selective serotonin reuptake inhibitors and psychotropic drugs have all been linked to dry eye.93
Conclusion
Dry eye is a complex multifactorial disease with symptoms ranging from mildly irritable to severely debilitating. ADDE is in itself a complex and multifactorial condition requiring careful questioning of patient symptoms, ocular health history, general health and medication use, family health and lifestyle to help guide the clinical exam to ascertain an accurate diagnosis. Patients with more serious underlying health concerns may first attend your practice with symptoms of dry eye, therefore it is important that as practitioners we are aware of the myriad of potential reasons for DED and how to identify them.
Keeping a clear and open mind, following a systematic but reactive examination process will better enable a clearer understanding of the route of a patients’ symptoms and therefore how best to treat them. ADDE and EDE should not be considered in isolation but as potentially a continuum or at the very least overlapping condition.
Part six of the series will look at examining the ADDE patient, the treatment options available and will include a case history highlighting both.
Craig McArthur is involved in a dedicated anterior eye clinic service at Peter Ivins Eyecare practice in Glasgow.
References
1.Mathers WD. Evaporation from the ocular surface. Exp Eye Res 2004;78: 389–394.
2.Mathers WD, lane Ja, Sutphin Je, Zimmerman MB. Model for ocular tear film function. Cornea 1996;15:110-9
3.Mathers WD, Daley Te. Tear flow and evaporation in patients with and without dry eye. Ophthalmology 1996;103:664-9
4.Mathers WD. Ocular evaporation in meibomian gland dysfunction and dry eye. Ophthalmology 1993;100:347-51
5.Yeh S, Song XJ, farley W, et al. apoptosis of ocular surface cells in experimentally induced dry eye. Invest Ophthalmol Vis Sci 2003;44:124-9De paiva cS, corrales rM, Villarreal al, et al. corticosteroid and doxycycline suppress MMp-9 and inflammatory cytokine expression, MapK activation in the corneal epithelium in experimental dry eye. Exp Eye Res 2006;83:526-35
6.Kunert KS, Tisdale aS, Gipson iK. Goblet cell numbers and epithelial proliferation in the conjunctiva of patients with dry eye syndrome treated with cyclosporine. Arch Ophthalmol 2002;120:330-7 218.
7.Brignole f, pisella pJ, Goldchild M, et al. flow cytometric analysis of inflammatory markers in conjunctival epithelial cells of patients with dry eyes. Invest Ophthalmol Vis Sci 2000;41:1356-63
8.Yokoi N, Takehisa Y, Kinoshita S. correlation of tear lipid layer interference patterns with the diagnosis and severity of dry eye. Am J Ophthalmol 1996;122:818-24
9.Kunert KS, Tisdale aS, Gipson iK. Goblet cell numbers and epithelial proliferation in the conjunctiva of patients with dry eye syndrome treated with cyclosporine. Arch Ophthalmol 2002;120:330-7
10.Fujihara T, Murakami T, Nagano T, et al. iNS365 suppresses loss of corneal epithelial integrity by secretion of mucin-like glycoprotein in a rabbit short-term dry eye model. J Ocul Pharmacol Ther 2002;18:363-70 (BS1)
11.Yerxa Br, Douglass JG, elena pp, et al. potency and duration of action of synthetic p2Y2 receptor agonists on Schirmer scores in rabbits Adv Exp Med Biol 2002;506(pt a):261-5 (BS2)
12.li DQ, chen Z, Song XJ, et al. Stimulation of matrix metalloproteinases by hyperosmolarity via a JNK pathway in human corneal epithelial cells. Invest Ophthalmol Vis Sci 2004;45:4302-11 57.
13.luo l, li DQ, corrales rM, pflugfelder Sc. hyperosmolar saline is a proinflammatory stress on the mouse ocular surface. Eye Contact Lens 2005;31:186-93
14.Tsubota K, Yamada M. Tear evaporation from the ocular surface. Invest Ophthalmol Vis Sci 1992;33:2942-50
15.Ousler GW 3rd, abelson MB, Nally la, et al: evaluation of time to ‘natural compensation’ in normal and dry eye subject populations during exposure to a controlled adverse environment. Adv Exp Med Biol 2002;506(pt B):1057-63
16.Moss Se, Klein r, Klein Be. prevalence of and risk factors for dry eye syndrome. Arch Ophthalmol 2000;118:1264-68
17.Nakamura h, Kawakamu a, Eguchi K. Mechanisms of autoantibody production and the relationship between autoantibodies and the clinical manifestations in Sjogren’s syndrome. Trans Res 2006;148(6):281-8
18.Hayashi Y, Arakaki r, Ishimaru N. The role of caspase cascade on the development of primary Sjögren’s syndrome. J Med Invest 2003;50:32-8
19.Zoukhri D. effect of inflammation on lacrimal gland function. Exp Eye Res 2006;82:885-98. [epub 2005 Nov 23]
20.Dawson l, Tobin a, Smith p, Gordon T. Antimuscarinic antibodies in Sjogren’s syndrome: where are we, and where are we going? Arthritis Rheum 2005;52:2984-95
21.Dawson lJ, Stanbury J, Venn N, et al. Antimuscarinic antibodies in primary Sjogren’s syndrome reversibly inhibit the mechanism of fluid secretion by human submandibular salivary acinar cells. Arthritis Rheum 2006;54:1165-73
22.Vitali c, Bombardieri S, Jonsson r, et al. classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American European consensus Group. Ann Rheum Dis 2002;61:554-8
23.Vitali c, Bombardieri S, Moutsopoulos hM, et al. Preliminary criteria for the classification of Sjögren’s syndrome. results of a prospective concerted action supported by the European community. Arthritis Rheum 1993;36:340-7
24.Fox RI, Robinson CA, Curd JG, et al. Sjögren’s syndrome. Proposed criteria for classification. Arthritis Rheum 1986;29:477-585
25.Toda i, Asano-Kato N, Komai-hori Y, Tsubota K. Dry eye after laser in situ keratomileusis. Am J Ophthalmol 2001;132:1-7
26.Wilson Se. laser in situ keratomileusis-induced (presumed) neurotrophic epitheliopathy. Ophthalmology 2001;108:1082-7
27.Hammond MD, Madigan Wp Jr, Bower KS. refractive surgery in the United States army, 2000-2003. Ophthalmology 2005;112:184-90
28.Battat l, Macri a, Dursum D, Pflugfelder Sc. effects of laser in situ keratomileusis on tear production, clearance, and the ocular surface. Ophthalmology 2001;108:1230-5
29.Pharmakakis NM, Katsimpris JM, Melachrinou Mp, Koliopoulos JX. Corneal complications following abuse of topical anesthetics. Eur J Ophthalmol 2002;12:373-8
30.Chen HT, Chen Kh, Hsu WM. Toxic keratopathy associated with abuse of low-dose anaesthetic: a case report. Cornea 2004;23:527-9
31.Vajdic C, Holden BA, et al. The frequency of ocular symptoms during spectacle and daily soft and rigid contact lens wear. Optom Vis Sci 1999;76:705-11
32.Begley CG, Caffery B, Nichols KK, Chalmers R. Responses of contact lens wearers to a dry eye survey. Optom Vis Sci 2000;77:40-6
33.Pritchard N, Fonn D, Brazeau D. Discontinuation of contact lens wear: a survey. Int Contact Lens Clin 1999;26:157-62
34.Richdale K, Sinnott lT, Skadahl e, Nichols JJ. frequency of and factors associated with contact lens dissatisfaction and discontinuation. Cornea 2007;26:168-74
35.Seedor Ja, lamberts D, Bergmann RB, Perry HD. Filamentary keratitis associated with diphenhydramine hydrochloride (Benadryl). Am J Ophthalmol 1986;101:376-7 (CS3)
36. Moss Se, Klein r, Klein Be. Prevalence of and risk factors for dry eye syndrome. Arch Ophthalmol 2000;118:1264-68
37.Mader Th, Stulting RD. Keratoconjunctivitis sicca caused by diphenoxylate hydrochloride with atropine sulfate (lomotil). Am J Ophthalmol 1991;111:377-8 (CS2)
38.Dart J. Cicatricial pemphigoid and dry eye. Semin Ophthalmol 2005;20: 95-100
39.Eschle-Meniconi Me, Ahmad Sr, foster CS. Mucous membrane pemphigoid: an update. Curr Opin Ophthalmol 2005;16:303-7
40.Hingorani M, Lightman S. Ocular Cicatricial pemphigoid. Curr Opin Allergy Clin Immunol 2006;6: 373-8
41.Tomlinson a, Khanal S. assessment of tear film dynamics: quantification approach. Ocul Surf 2005;3:81-95
42.Craig JP, Tomlinson A. Age and gender effects on the normal tear film. Adv Exp Med Biol 1998; 438:411-5
43.Shimazaki J, Sakata M, Tsubota K. Ocular surface changes and discomfort in patients with meibomian gland dysfunction. Arch Ophthalmol 1995;113:1266-70
44.Rolando M, Refojo Mf, Kenyon Kr. increased tear evaporation in eyes with keratoconjunctivitis sicca. Arch Ophthalmol 1983;101:557-8
45.Rolando M, Refojo Mf, Kenyon Kr. Tear water evaporation and eye surface diseases. Ophthalmologica 1985;190:147-9
46.Tsubota K, Yamada M. Tear evaporation from the ocular surface. Invest Ophthalmol Vis Sci 1992;33:2942-50
47.Goebbels M. Tear secretion and tear film function in insulin dependent diabetics. Br J Ophthalmol 2000;84:19-21
48.Mathers WD, lane Ja, Sutphin Je, Zimmerman MB. Model for ocular tear film function. Cornea 1996;15:110-9
49.Fox RI. Sjögren’s syndrome. Lancet. 2005;366:321–331.
50.Hayashi Y, Arakaki r, Ishimaru N. The role of caspase cascade on the development of primary Sjogren’s syndrome. J Med Invest 2003;50:32-8
51.Zoukhri D. effect of inflammation on lacrimal gland function. Exp Eye Res 2006;82:885-98. [epub 2005 Nov 23]
52.Dawson l, Tobin a, Smith p, Gordon T. Antimuscarinic antibodies in Sjogren’s syndrome: where are we, and where are we going? Arthritis Rheum 2005;52:2984-95
53.Dawson lJ, Stanbury J, Venn N, et al. Antimuscarinic antibodies in primary Sjogren’s syndrome reversibly inhibit the mechanism of fluid secretion by human submandibular salivary acinar cells. Arthritis Rheum 2006;54:1165-73
54.Wiik A, Cervera R, Haass M, et al. European attempts to set guidelines for improving diagnostics of autoimmune rheumatic disorders. Lupus 2006;15:391-6
55.Pflugfelder SC, Huang AJ, Feuer W, et al. Conjunctival cytologic features of primary Sjögren’s syndrome. Ophthalmology. 1990;97:985–991.
56.Tsubota K, Toda I, Yagi Y, et al. Three different types of dry eye syndrome. Cornea. 1994;13:202–209.
57.Xu KP, Katagiri S, Takeuchi T, et al. Biopsy of labial salivary glands and lacrimal glands in the diagnosis of Sjögren’s syndrome. J Rheumatol. 1996;23:76–82
58.Mathers WD, lane Ja, Zimmerman MB. Tear film changes associated with normal aging. Cornea 1996;15: 229-34
59.Sahlin S, chen e. evaluation of the lacrimal drainage function by the drop test. Am J Ophthalmol 1996;122:701-8
60.Tomlinson A, Geisbrecht J. The aging tear film. Br J Contact Lens Assoc 1993;16;67-9
61.Norn MS. Semi-quantitative interference study of the fatty layer of the pre-corneal film. Acta Ophthalmol (Copenh) 1979;57:766-74
62.Obata h, Yamamoto S, Horiuchi H, Machinami R. Histopathologic study of human lacrimal gland. Statistical analysis with special reference to aging. Ophthalmology 1995;102:678-86
63.Damato Be, Allan D, Murray SB, lee Wr. Senile atrophy of the human lacrimal gland: the contribution of chronic inflammatory disease. Br J Ophthalmol 1984;68:674-80
64.Davidoff E, Friedman AH. Congenital Alacrima. Surv Ophthalmol 1977;22:113-9
65.Arya SK, Chaudhuri Z, Jain r, et al. Congenital alacrima in pierre robin sequence. Cornea. 2004; 23:632-4
66.Brooks Bp, Kleta r, Stuart c, et al. Genotypic heterogeneity and clinical phenotype in triple a syndrome: a review of the Nih experience 20002005. Clin Genet 2005;68:215-21
67.Krumbholz M, Koehler K, Huebner A. Cellular localization of 17 natural mutant variants of alaDiN protein in triple a syndrome—shedding light on an unexpected splice mutation. Biochem Cell Biol 2006;84:243-9
68.Axelrod FB, Chelimsky GG, Weese-Mayer De. Paediatric autonomic disorders. Pediatrics 2006;118: 309-21
69.Gold-von Simson G, Axelrod FB. Familial dysautonomia: update and recent advances. Curr Probl Pediatr Adolesc Health Care 2006;36:218-37
70.James DG, Anderson R, Langley D, Ainslie D. Ocular sarcoidosis. Br J Ophthalmol 1964;48:461-70
71.Heath P. Ocular lymphomas. Trans Am Ophthalmol Soc 1948;46:385-98
72.Itescu S, Brancato lJ, Buxbaum J, et al. A diffuse infiltrative cD8 lymphocytosis syndrome in human immunodeficiency virus (hiV) infection: a host immune response associated with hla-Dr5. Ann Intern Med 1990;112:3-10
73.Ogawa Y, Okamoto S, Wakui M, et al. Dry eye after haematopoietic stem cell transplantation. Br J Ophthalmol 1999;83:1125-30
74.Ogawa Y, Kuwana M, Yamazaki K, et al. periductal area as the primary site for T-cell activation in lacrimal gland chronic graft- versus-host disease. Invest Ophthalmol Vis Sci 2003;44:1888-96
75.Scherz W, Dohlman ch. Is the lacrimal gland dispensable? Keratoconjunctivitis sicca after lacrimal gland removal. Arch Ophthalmol 1975;93: 81-3
76.Whitwell J. Denervation of the lacrimal gland. Br J Ophthalmol 1958;42:518-25
77.Guzey M, Ozardali I, Basar E, et al. A survey of trachoma: the histopathology and the mechanism of progressive cicatrization of eyelid tissues. Ophthalmologica 2000;214: 277-84
78.Dart J. Cicatricial pemphigoid and dry eye. Semin Ophthalmol 2005;20: 95-100
79.Eschle-Meniconi ME, Ahmad SR, Foster CS. Mucous membrane pemphigoid: an update. Curr Opin Ophthalmol 2005;16:303-7
80.Hingorani M, Lightman S. Ocular cicatricial pemphigoid. Curr Opin Allergy Clin Immunol 2006;6: 373-8
81.Power WJ, Ghoraishi M, Merayo-lloves J, et al. Analysis of the acute ophthalmic manifestations of the erythema multiforme/Stevens-Johnson syndrome/toxic epidermal necrolysis disease spectrum. Ophthalmology 1995;102:1669-76
82.Lemp MA. Basic principles and classification of dry eye disorders, in Lemp MA, Marquandt R (eds). The dry eye: a comprehensive guide. New York, Springer,1992,pp 101-31
83.Farris RL, Stuchell RN, Mandel ID. Tear osmolarity variation in the dry eye. Trans Am Ophthalmol Soc 1986;84:250-68
84.De Paiva CS, Pflugfelder SC. Corneal epitheliopathy of dry eye induces hyperesthesia to mechanical air jet stimulation. Am J Ophthalmol 2004;137: 109-15
85.Seifart U, Strempel I. The dry eye and diabetes mellitus. Ophthalmologe 1994;91:235-9
86.Moss Se, Klein r, Klein BE. Prevalence of and risk factors for dry eye syndrome. Arch Ophthalmol 2000;118:1264-8
87.Moss Se, Klein r, Klein Be. incidence of dry eye in an older population. Arch Ophthalmol 2004; 122: 369-73
88.Kaiserman I, Kaiserman N, Nakar S, Vinker S. Dry eye in diabetic patients. Am J Ophthalmol 2005;139: 498-503
89.Cavanagh HD, Colley AM. The molecular basis of neurotrophic keratitis. Acta Ophthalmol Suppl 1989;192:115-34
90.Cavanagh HD, Colley AM. The molecular basis of neurotrophic keratitis. Acta Ophthalmol Suppl 1989;192:115-34
91.Jordan A, Baum J. Basic tear flow. Does it exist? Ophthalmology 1980;87:920.
92.Baum JL, Adler Me. Pheochromocytoma, medullary thyroid carcinoma, multiple mucosal neuroma. a variant of the syndrome. Arch Ophthalmol 1972;87: 74-84
93.Moss SE, Klein R, Klein BE. Incidence of dry eye in an older population. Arch Ophthalmol 2004; 122: 369-73