Retinoblastoma is by far the commonest eye cancer of childhood. It fortunately is a rare disease but with significant morbidity and can also carry a very significant risk to life depending where on the planet children are affected by the condition.
Epidemiology
The incidence in Europe is 2.7 to 3.3 per million in 0-14 year olds1 and, because the onset is commoner in younger children, the rate is even higher in younger babies and was measured at 65 per million live births in a 2004 Scandinavian survey.2 There is recent evidence that the rate has increased a little in Europe to 72 per million live births.3 This might be because of improved survival of people with retinoblastoma to have their own children with retinoblastoma.
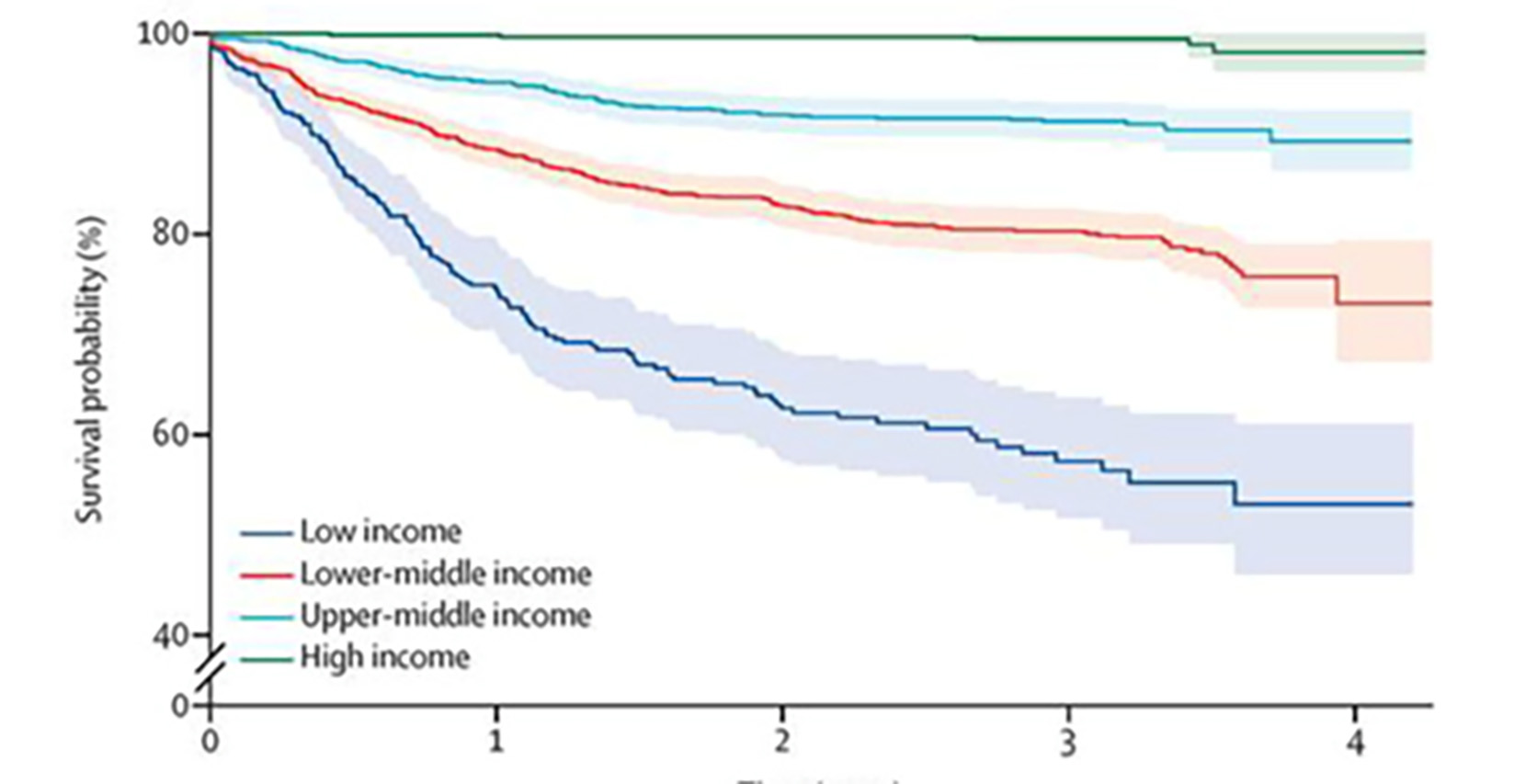
In a 2017 global survey from 149 countries,4 it was demonstrated that survival varies enormously depending on the wealth of the country in which a child presents with the disease. This outcome disparity remains one of the most important challenges facing children’s eye care internationally. Three-year survival in high income countries was 98.8-100% (95% confidence interval), 89.5-93.0% for upper-middle-income countries, 78.3-82.3% for lower-middle income countries and 52.1-63.0% for low income countries (figure 1).
Figure 1: Relationship between income and survival rate from retinoblastoma4
Inheritance
The genetic origin for nearly all retinoblastomas is due to the retinal cell having defective versions of both of its copies of the RB1 gene and therefore not able to produce effective amounts of pRB; a protein that prevents tumour formation. We normally have two functional copies of RB1 in each cell; one on each copy of chromosome 13. If these are both non-functional, that is if two ‘hits’ have occurred, then a tumour usually is the result.
The reason that these two ‘hits’ accrue differs between individuals and reflects the heritability patterns of retinoblastoma of which there are several forms as follows:5
- Familial heritable form: A parental dysfunctional RB1 gene is inherited in all cells in the body and every cell has a predisposition to tumour formation, but in particular the retinal cells in infancy. The parental copy of RB1 is the first ‘hit’. When the second ‘hit’ is acquired in a cell it forms goes on to form a tumour. This form is characterised by a family history and early onset disease, usually bilateral with multiple tumours. There is a lifelong increased risk of ‘second primary’ cancers. Affected people pass on their tendency to retinoblastoma to half of their children.
- Isolated heritable form: A de-novo dysfunctional change occurs in one copy of the RB1 gene at a very early (pre-zygotic) phase of embryo development so the first ‘hit’ is in every cell in the individual but is not found widespread in either parents’ body. When the second ‘hit’ occurs in a cell a tumour can result. This form is clinically identical to the familial form except there is no family history of disease.
- Non-heritable form: Originally both copies of the RB1 gene are functional in all the cells in the body but then both ‘hits’ occur in a retinal cell leading to uncontrolled proliferation and a tumour. This form is characterised by no family history, unilateral disease, slightly older presentation, no increased lifelong risk of cancer formation and no tendency to pass on the disease.
- Mosaic form: If a mutation occurs in a cell soon after formation of the zygote then not all cells carry the RB1 mutation but all the descendent cells have an RB1 mutation. The tendency to tumour formation depends upon what proportion and which parts of the body are affected so differs from one individual to another. It is usually unilateral. It can pass on to the next generation if the sperm or egg cells of the affected individual have the mutation.
Recent publications have shown that socioeconomic circumstance is related to the relative prevalence of the different inheritability patterns (familial heritable vs non-heritable) of retinoblastoma. Low-income countries have a relatively higher proportion of non-heritable disease.6
This is probably because in low-income countries survival from heritable disease is worse and those people have a smaller chance of surviving to have children of their own in the next generation with retinoblastoma.
Diagnosis
Retinoblastoma remains one of the few areas in clinical medicine in which a diagnosis of cancer is made without laboratory confirmation from a tissue sample. It is not safe, in most instances, to take a traditional biopsy, which would allow histological confirmation; that is to remove a part of the solid element of the tumour and to look at it under a microscope. To do so would allow tumour cells to escape the eye along the track of the instruments used to take the sample.
Furthermore, a diagnosis has to be made quickly to avoid disease progression and the associated increased risks while awaiting genetic absolute confirmation.
Of note, there has been significant progress with the genetic investigation of retinoblastoma. For many years geneticists have been able to find mutations in the retinoblastoma gene either from DNA in blood from a child with the disease or from the tumour itself if the eye had been removed along with the tumour. These tests take a month or so to get results.
Recently in the UK, pregnant mothers have been able to find out whether their unborn baby carries a mutation in the RB gene just from a non-invasive blood test from the arm (non-invasive pre-natal diagnosis or NIPD), rather than from tests in which material is taken from within the womb while the mother-to-be is pregnant; a development that avoids the risks of more invasive tests (chorionic villus sampling or amniocentesis) to the unborn baby.7 However, this is a new process and not available to everyone. For example, if the mother has heritable Rb, she can only have NIPD if she already has a child. Find out more at https://chect.org.uk/new-pregnancy-testing-technology-now-available/.
Also, it has recently become possible to find out which gene has gone wrong from a fluid sample from the eye, without surgically removing the eye.8, 9 This is known as an aqueous tap or fluid biopsy. Since the enormous improvements in outcome for retinoblastoma over the past 10 years especially for group D eyes, so many more eyes have been saved and those cases of unilateral, non-germline disease will likely benefit most from this means of investigation, which will be the best chance to know the genetic ‘signature’ of the tumour that will not be present elsewhere in the child’s body.
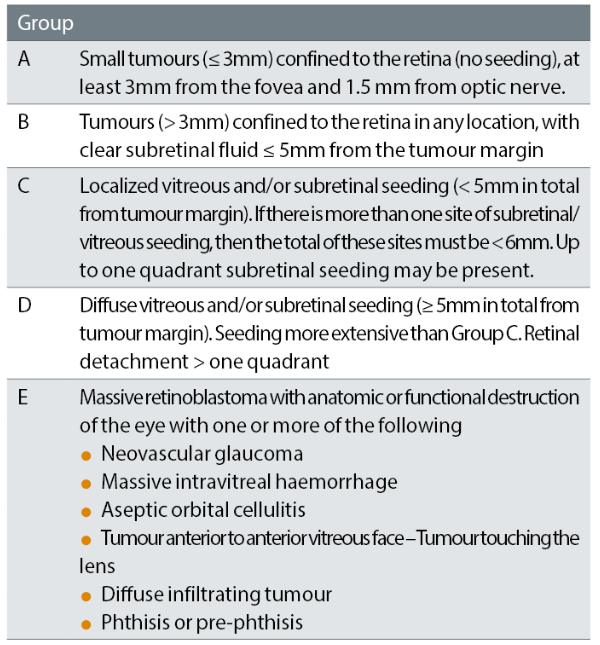
Retinoblastomas have been categorised according to the definitions of the International Intraocular Retinoblastoma Classification (IIRC) for many years. This describes the severity of the features of disease at presentation and remains in common use.
The categories of disease are A to E with ‘A’ being the least severe, ‘E’ being the most severe (table 1).
Table 1: International intraocular retinoblastoma classification (IIRC)10
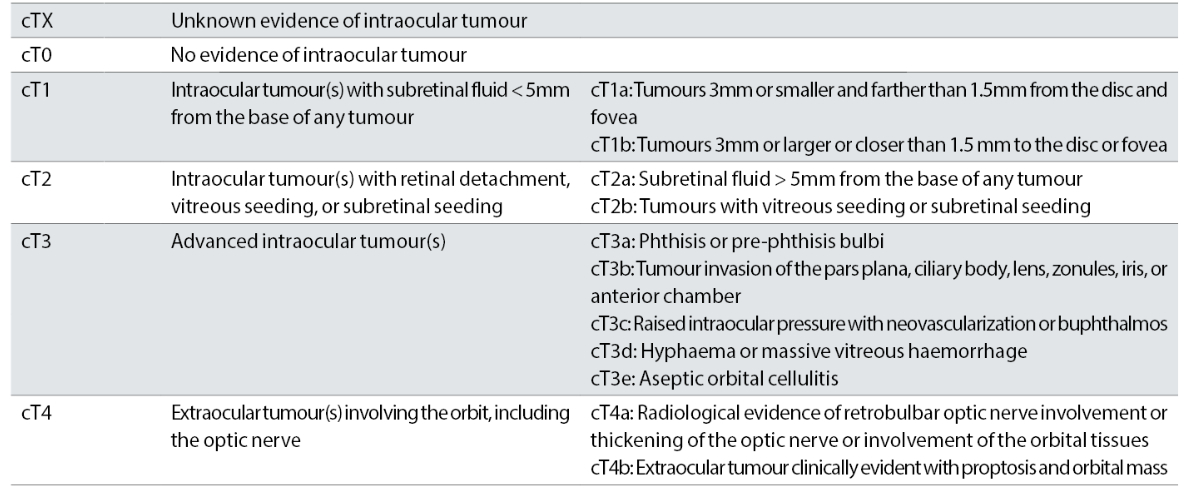
The more recent classification (AJCC) is more comprehensive and complex and takes into account important factors including heritability. This is a fundamentally important descriptor since it will stratify the risk of new tumours, in particular in younger children and reflects the lifelong risk of ‘second primary’ tumours for those people who carry a mutation in their RB1 gene and hence a predisposition to new cancers (table 2).
Table 2: Definitions for 8th Edition AJCC clinical primary tumour staging of retinoblastoma, part of larger TNMH staging schema including pathology and heritability descriptors11
Both the IIRC and AJCC classifications are used in correspondence describing children’s retinoblastoma among eye care professionals.
The AJCC pathological descriptors of retinoblastoma highlight the importance of diagnosis and intervention before the tumour has had a chance to get extend beyond the anatomical barriers of the eye. With each successive breach the treatment is escalated to reflect the increased risk of mortality and extraocular metastasis.
Taking the example of disease along the optic nerve; if disease is contained within the laminar cribrosa (pT2) no adjuvant chemotherapy is required, post-laminar optic nerve involvement (PLONI, pT3) indicates systemic etoposide, carboplatin and vincristine and cut-end disease of the optic nerve (at enucleation) means disease beyond the globe (pT4) and escalation to consider proton treatment and a marked escalation of chemotherapy/oncology treatment.
The exact approach to these scenarios is yet to be informed by high quality prospective evidence so practice varies between centres and countries but the underlying principle is agreed.
Referral
Despite all the exciting steps forward in treating retinoblastoma, one of the most important steps in achieving a good outcome remains early detection and treatment. Awareness campaigns have had a measurable shortening time to diagnosis12 and can reduce the proportion of children with extraocular spread at diagnosis.13 In the UK, the Childhood Eye Cancer Trust (CHECT) is an excellent source of educational material (chect.org.uk).
Retinoblastoma can be noticed by their parents because tumours can cause:
- Strabismus
- Reduced vision
- Red or painful eye
Less commonly, parents notice a white pupil, known as leukocoria, or a white pupil on flash photography, known as photoleukocoria (figures 3 and 4).
Figure 3: Examples of leukocoria

It is of great importance that optometrists and primary care doctors can recognise children who might have a retinoblastoma among the large number of eye complaints they diagnose and treat.
The hallmark of the examination, which can identify children at risk, is the red reflex examination. This is the inspection of the reflection of light from the child’s retina and can most effectively be done with a direct ophthalmoscope. The best way to do this is to use a well charged ophthalmoscope in a dark room and is better facilitated by using eye drops to pharmacologically dilate the pupils.
It is important that this test is done as part of every child’s eye examination. Depending on the expertise of the examiner, fundoscopy is also very useful to visualise the retinal features. If there are concerns about a child having a retinoblastoma, an urgent referral to a paediatric ophthalmology service should be undertaken immediately.
If the paediatric ophthalmologist confirms suspicious findings, an urgent referral should be made to one of the two national centres in the UK; the Royal London Hospital or the Birmingham Children’s Hospital.
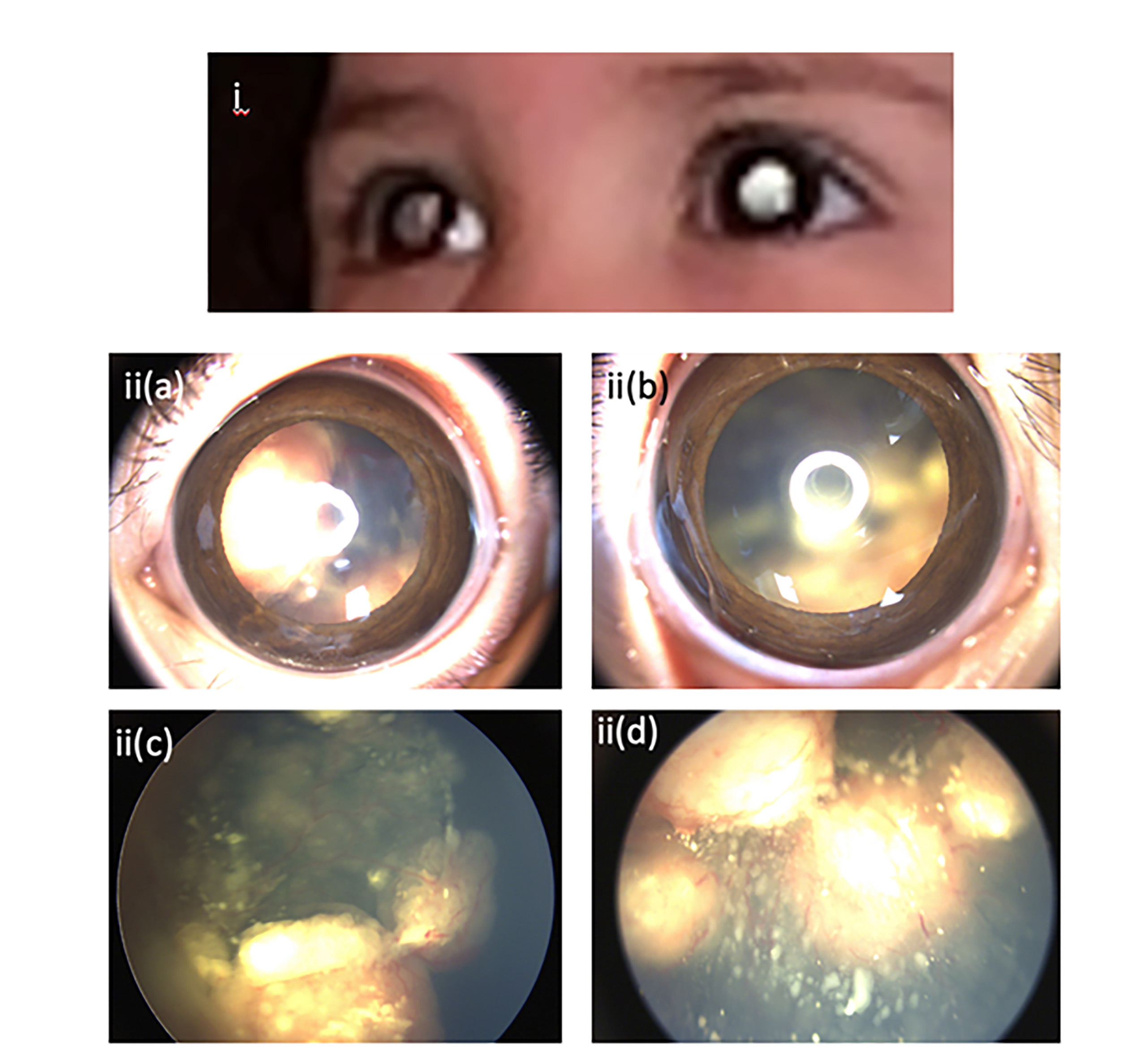
Unfortunately, even in high-income economies, we still see delayed referrals (figure 4), typically after a red eye is thought to have been conjunctivitis or a strabismus is thought to be due to an innocent cause, and the great majority of these children can be easily identified by a red reflex examination done by a practitioner familiar with the test.
Figure 4: Parentally witnessed leukocoria and photo-leukocoria (i). RetCam images of advanced bilateral group D cT2b retinoblastoma before treatment: ii(a), ii(b); right and left eye external appearance, ii(c), ii(d); right and left retinal appearance. The child had contact with four eye care professionals electively who did not suggest the diagnosis until six months later when the family attended emergency services resulting in a diagnosis from a paediatric ophthalmologist (images shared with parental permission)
There are ongoing campaigns around the world and organisations which exist to raise awareness of retinoblastoma and enhance early detection, including those of the European Retinoblastoma Group (EuRBG), the Rétinostop association in France, and the Childhood Eye Cancer Trust (CHECT) in the UK.
Update on treatment
In the late 20th century, the treatments for retinoblastoma were principally:
- Enucleation; removing the entire globe and replacing this with an artificial implant in the eye socket, with an overlying cosmetic ‘shell’
- Systemic chemotherapy; with vincristine, carboplatin and etoposide, known as ‘JOE’ chemotherapy
- Laser
- Cryo-therapy; freeze treatment applied through the wall of the eye
In some cases, local radiation brachytherapy with a radioactive plaque emitting radiation that penetrates very short distances was, and is still, a good treatment in certain cases. All of these 20th century therapies continue to play an important part in retinoblastoma treatment.
External beam radiotherapy (EBRT) fell rapidly from use in all but the most extreme circumstances when it was realised that, in particular for people with an RB1 mutation, EBRT increases the risk of later malignancies and death.15
In the 21st century, new treatments have dramatically improved outcomes for advanced intra-ocular tumours and in particular group D, cT2b disease. Intravitreal chemotherapy, popularised and optimised by Professor Munier from Lausanne, has allowed treatment of disease seeding into the vitreous, which, previously, in many cases would have obligated removal of the eye.
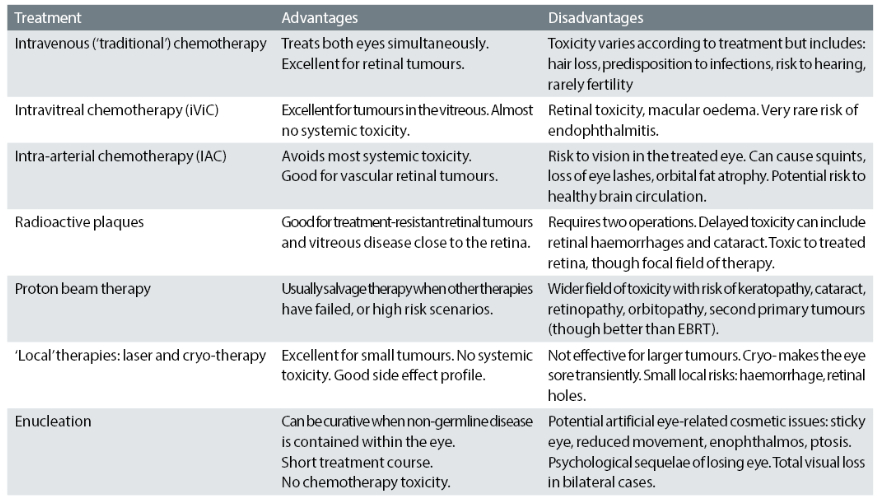
With time, the indications for intra-vitreal chemotherapy have widened and refinement of the dose has further expanded the utility of this treatment option.17 Table 3 summarises the common treatment modalities and selected relative advantages and disadvantages.
Table 3: Common treatment modalities and selected relative advantages and disadvantages
The aims of the management of retinoblastoma are, most importantly, to protect the child’s life from the tumour and, where possible, to prevent them losing an eye; and, of course, with the aim to preserve the vision.
Children suspected of having an eye tumour are examined under anaesthesia to assess their condition. They are assessed with:
- Tonometry
- Slit lamp anterior segment examination
- Indirect ophthalmoscopy
- Retinal imaging
- Ultrasound (including high frequency ‘biomicroscopy’)
- Optic coherence tomography
- Fluorescein angiography (occasionally)
Subsequently, most children have magnetic resonance imaging (MRI) to assess the degree of optic nerve or brain involvement. Every child has a treatment plan designed to best treat their retinoblastoma/s after discussion with the family and a multi-disciplinary team, including oncologists and ophthalmologists with expertise in the disease.
Clearly these are life-changing discussions for parents, which must be given the sensitivity and time they need. A team approach, with support from trained counsellors and mental health professionals, can help support families through these difficult periods.
Summary
The last 20 years have seen huge advances in retinoblastoma care; in particular, genetic investigation of the disease and therapeutic options. We can reasonably expect further advances in both areas in more affluent countries, as cell-free DNA analysis techniques expand to allow diagnosis (including pre-natal), prognostication and personalised targeted treatment informed by a genetic understanding of a given tumour. Survival is excellent for retinoblastoma in high income countries and the drive for improving outcomes is to achieve even better rates of preservation and vision.
In low-income countries, mortality is unacceptably high and overcoming the barriers to equitable outcomes for children in less affluent global regions remains a significant challenge to overcome in the next 20 years.
- Mr Joe Abbott is a consultant paediatric ophthalmologist at Birmingham Children’s Hospital.
References
- Gatta G, Ferrari A, Stiller CA, Pastore G, Bisogno G, Trama A, Capocaccia R; RARECARE Working Group. Embryonal cancers in Europe. European Journal of Cancer. 2012 Jul;48(10):1425-33. doi: 10.1016/j.ejca.2011.12.027. Epub 2012 Feb 20. PMID: 22357215
- Seregard S, Lundell G, Svedberg H, Kivelä T. Incidence of retinoblastoma from 1958 to 1998 in Northern Europe: advantages of birth cohort analysis. Ophthalmology. 2004 Jun;111(6):1228-32. doi: 10.1016/j.ophtha.2003.10.023. PMID: 15177976
- Stacey, Andrew W.Harby, Lamis Al et al. Incidence of Retinoblastoma Has Increased: Results from 40 European Countries. Ophthalmology, Volume 128, Issue 9, 1369 – 1371
- Global Retinoblastoma Study Group. The Global Retinoblastoma Outcome Study: a prospective, cluster-based analysis of 4064 patients from 149 countries. Lancet Glob Health. 2022 Aug;10(8):e1128-e1140. doi: 10.1016/S2214-109X(22)00250-9. PMID: 35839812; PMCID: PMC9397647
- Munier, F. L. et al. Conservative management of retinoblastoma_ Challenging orthodoxy without compromising the state of metastatic grace. “Alive, with good vision and no comorbidity.” Progressions in Retinal Eye Research, 73, 100764 (2019)
- Global Retinoblastoma Study Group; Fabian ID, et al. Global Retinoblastoma Presentation and Analysis by National Income Level. JAMA Oncology, 2020 May 1;6(5):685-695. doi: 10.1001/jamaoncol.2019.6716. Erratum in: JAMA Oncol. 2020 Nov 1;6(11):1815. PMID: 32105305; PMCID: PMC7047856
- Gerrish A, Bowns B, Mashayamombe-Wolfgarten C, Young E, Court S, Bott J, McCalla M, Ramsden S, Parks M, Goudie D, Carless S, Clokie S, Cole T, Allen S. Non-Invasive Prenatal Diagnosis of Retinoblastoma Inheritance by Combined Targeted Sequencing Strategies. Journal of Clinical Medicine, 2020 Oct 30;9(11):3517. doi: 10.3390/jcm9113517. PMID: 33143217; PMCID: PMC7692133
- Berry JL, Xu L, Murphree AL, Krishnan S, Stachelek K, Zolfaghari E, McGovern K, Lee TC, Carlsson A, Kuhn P, Kim JW, Cobrinik D, Hicks J. Potential of Aqueous Humor as a Surrogate Tumor Biopsy for Retinoblastoma. JAMA Ophthalmology, 2017 Nov 1;135(11):1221-1230. doi: 10.1001/jamaophthalmol.2017.4097. PMID: 29049475; PMCID: PMC5710399
- Gerrish A, Stone E, Clokie S, Ainsworth JR, Jenkinson H, McCalla M, Hitchcott C, Colmenero I, Allen S, Parulekar M, Cole T. Non-invasive diagnosis of retinoblastoma using cell-free DNA from aqueous humour. British Journal of Ophthalmology, 2019 Feb 11;103(5):721–4. doi: 10.1136/bjophthalmol-2018-313005
- Linn Murphree A. Intraocular retinoblastoma: the case for a new group classification. Ophthalmology Clinic of North America, 2005; 18(1):41–54, viii
- Mallipatna, A.C et al. Retinoblastoma. In: Amin MB, Edge SB, Greene FL, Byrd DR, Brookland RK, Washington MK et al., editors. AJCC Cancer Staging Manual, 8th ed. New York: Springer; 2017.0.819–31
- Wallach, M., Balmer, A., Munier, F., Houghton, S., Pampallona, S., von der Weid, N., Beck-Popovic, M., Swiss Pediatric Oncology, G., Swiss Childhood Cancer, R., 2006. Shorter time to diagnosis and improved stage at presentation in Swiss patients with retinoblastoma treated from 1963 to 2004. Pediatrics, 118, e1493–1498
- Leander, C., Fu, L.C., Pena, A., Howard, S.C., Rodriguez-Galindo, C., Wilimas, J.A., Ribeiro, R.C., Haik, B., 2007. Impact of an education program on late diagnosis of retinoblastoma in Honduras. Pediatric Blood Cancer, 49, 817–819
- Gurney, S. P., Makanjuola, T., Kutubi, Muhammad, Parulekar, M. & Abbott, J. How to use…the direct ophthalmoscope. Archives of Disability and Child Educational Practice. Ed 103, 102–109 (2018)
- Moll, A. C., Imhof, S. M., Schouten-Van Meeteren, A. Y., Kuik, D. J., Hofman, P., & Boers, M. (2001). Second primary tumors in hereditary retinoblastoma: a register-based study, 1945-1997: is there an age effect on radiation-related risk? Ophthalmology, 108(6), 1109–1114
- Munier, F.L., Soliman, S., Moulin, A.P., Gaillard, M.C., Balmer, A., Beck-Popovic, M., 2012b. Profiling safety of intravitreal injections for retinoblastoma using an antireflux procedure and sterilisation of the needle track. British Journal of Ophthalmology, 96, 1084–1087
- Abramson, D. H. & Francis, J. H. Intravitreal topotecan 90 µg for recurrent solid retinoblastoma tumors is effective and not toxic. Journal of Pediatric Ophthalmology & Strabismus, 60, e16–e18 (2023)